Ингибиторы янус-киназ: классификация, современное состояние и фармакологические свойства
Ингибиторы янус-киназ: классификация, современное состояние и фармакологические свойства

Аннотация
Обзор посвящен современному состоянию исследований в области низкомолекулярных ингибиторов янус-киназ (JAK) — перспективному классу таргетных противовоспалительных и противоопухолевых препаратов, известных как «якинибы». Янус-киназы (JAK1, JAK2, JAK3, TYK2) являются ключевыми цитоплазматическими тирозинкиназами, опосредующими передачу внутриклеточных сигналов от широкого спектра цитокинов и факторов роста через путь JAK/STAT. Гиперактивация этого пути лежит в основе патогенеза множества аутоиммунных, воспалительных и онкологических заболеваний, что делает янус-киназы привлекательными мишенями для фармакологического воздействия. В работе рассмотрены структурные особенности ферментов семейства JAK. Особое внимание уделено анализу классификации ингибиторов, основанной на механизме действия и селективности. Выделены обратимые АТФ-конкурентные ингибиторы I типа (тофацитиниб, барицитиниб, руксолитиниб), II типа (CHZ868, BBT594), а также аллостерические (деукравацитиниб, LS104, ON044580) и необратимые (ковалентные) ингибиторы (ритлецитиниб и др.). Для каждого класса приведены представители, детально описаны их молекулярные механизмы взаимодействия с ферментом, основные пути синтеза и современные области клинического применения. Систематизированы данные о фармакологических свойствах препаратов первого и второго поколений, подчеркивается эволюция в сторону повышения селективности и профиля безопасности. Отдельный раздел посвящен анализу побочных эффектов якинибов, связанных с иммуносупрессией, таких как риск инфекций, гематологические и биохимические изменения. Сделан вывод о высокой терапевтической значимости ингибиторов JAK, обеспечивающих быстрый и эффективный контроль над тяжелыми хроническими заболеваниями.
1. Введение
Семейство янус-киназ (Janus kinases, JAK) представляет собой группу внутриклеточных нерецепторных тирозинкиназ, играющих фундаментальную роль в передаче сигналов от широкого спектра цитокинов, интерферонов и факторов роста. Будучи ключевыми медиаторами сигнального пути JAK/STAT (Janus kinase / Signal Transducer and Activator of Transcription), эти ферменты регулируют каскад биохимических реакций, приводящих к активации транскрипции генов, ответственных за пролиферацию, дифференцировку, выживаемость клеток и активацию иммунного ответа. Физиологическая роль JAK (включая изоформы JAK1, JAK2, JAK3 и TYK2) охватывает такие жизненно важные процессы, как гемопоэз, лимфопоэз и поддержание иммунного гомеостаза.
Нарушение активности янус-киназ, вызванное мутациями, конститутивной активацией или патологически повышенной экспрессией цитокинов, напрямую связана с патогенезом целого ряда тяжелых и социально значимых заболеваний. К их числу относятся аутоиммунные и хронические воспалительные заболевания (ревматоидный артрит, псориатический артрит, воспалительные заболевания кишечника, атопический дерматит, псориаз), а также онкогематологические патологии (миелопролиферативные новообразования, некоторые виды лейкозов). Данное обстоятельство определило JAK в качестве терапевтически значимой мишени для фармакологического вмешательства.
Разработка низкомолекулярных ингибиторов янус-киназ, обозначаемых общим термином «якинибы», стала важным достижением в области таргетной терапии. Первое поколение этих препаратов (например, тофацитиниб, руксолитиниб) продемонстрировало высокую клиническую эффективность, доказав принципиальную возможность контролировать заболевание через модуляцию внутриклеточной передачи сигнала. Однако неселективный характер их действия, направленный одновременно на несколько изоформ JAK, зачастую был сопряжен с развитием побочных эффектов.
Дальнейшая эволюция в этой области привела к созданию второго поколения якинибов — селективных ингибиторов (филготиниб, упадацитиниб, аброцитиниб и др.), обладающих повышенной специфичностью к отдельным изоформам (преимущественно JAK1). Это позволило улучшить профиль безопасности, сохранив терапевтическую эффективность. Параллельно развиваются инновационные подходы к ингибированию JAK, включая аллостерические (деукравацитиниб, нацеленный на псевдокиназный домен TYK2) и необратимые (ковалентные) ингибиторы, открывающие новые возможности для лечения резистентных форм заболеваний.
В связи с высокой распространенностью патологий, в основе которых лежит гиперактивация пути JAK/STAT, непрерывный поиск и изучение новых ингибиторов остается одной из приоритетных задач современной фармакологии и медицины. Важное значение имеют не только исследования клинической эффективности и безопасности, но и глубокий анализ структуры этих соединений, механизмов их взаимодействия с мишенью, а также разработка эффективных синтетических методов их получения.
Целью данной обзорной статьи является систематизация современных данных о классификации, механизмах действия, фармакологических свойствах, методах синтеза и клиническом применении ингибиторов янус-киназ. Особое внимание уделяется эволюции от неселективных к высокоселективным и аллостерическим ингибиторам, что отражает магистральное направление развития этого класса лекарственных средств в сторону повышения эффективности и безопасности терапии.
2. Строение янус-киназ
Янус-киназы были идентифицированы в начале 1990-х годов. В семейство янус-киназ входят четыре киназы: JAK1, JAK2, JAK3, TYK2 (известная как тирозинкиназа Т). Киназы JAK1, JAK2 и TYK2 экспрессируются повсеместно, в то время как JAK3 ограничивается по большей части гемопоэтическими клетками .
Каждая янус-киназа выполняет определённые функции в клетке: JAK1 участвует в передаче сигналов от различных провоспалительных цитокинов, кроме этого, JAK1 и JAK3 принимают участие в лимфопоэзе, JAK2 передает сигналы от гемопоэтических факторов роста, а JAK3 и TYK2 участвуют в регуляции иммунной системы.
В структуру янус-киназ входят около 1100 аминокислотных остатков, которые распределяются между четырьмя функциональными доменами . Кроме киназного и псевдокиназного доменов, в структуру янус-киназ входят домены SH2 и FERM (см. рис. 1).
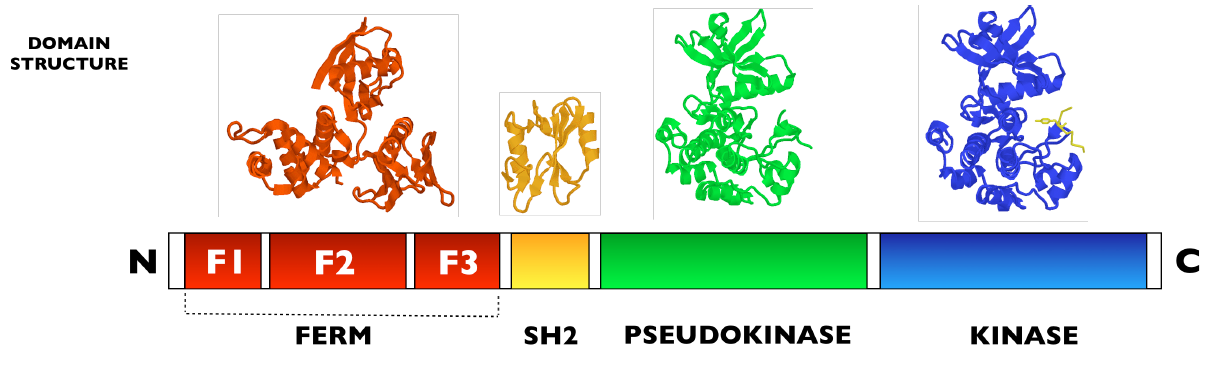
Рисунок 1 - Структура янус-киназ
Примечание: по ист. [3]
Домен FERM расположен в N-концевой доле, его основная функция — связывание с рецепторами цитокинов. В результате данного связывания образуется комплекс, аналогичный рецепторной тирозинкиназе. Домен FERM содержит приблизительно 400 аминокислотных остатков и состоит из трех субдоменов, расположенных в виде клевера. N-концевой субдомен F1 имеет убиквитин-подобную β-складку, субдомен F2 содержит множество α-спиралей и демонстрирует структурное сходство с ацетил-КоА-связывающим белком, а субдомен F3 имеет PH-домен (гомология плекстрина) .
Взаимодействие FERM и рецептора дополнительно способствует регуляции каталитической активности. Помимо этого, мутации домена FERM могут препятствовать фосфорилированию киназы и ее активации.
Домен SH2, следующий за доменом FERM, принимает участие в агрегации рецепторных комплексов на мембране и кросс-фосфорилировании. Домен SH2 образовывает связи со специфичными мотивами, содержащими фосфорилированные остатки тирозина. Он содержит примерно 100 аминокислот и состоит из двух α-спиралей и центрального антипараллельного β-структурного участка.
Домен псевдокиназы расположен между доменом SH2 и доменом киназы. Он состоит из приблизительно 300 аминокислотных остатков и структурно схож со стандартными каталитическими доменами, но в домене псевдокиназы отсутствуют консервативные остатки необходимые для киназной активности. Отсутствие возможности проявления каталитической активности связано с отсутствием остатка аспартата, который необходим для корректной ориентации АТФ в центре связывания .
Из 538 идентифицированных протеинкиназ человека 48 имеют псевдокиназные домены. Одновременное нахождение псевдокиназного и киназного домена в одной протеинкиназе отмечено только у пяти представителей данной группы. Это JAK1, JAK2, JAK3, TYK2 и протеинкиназа Gcn2, фосфорилирующая спиртовые группы серина и треонина.
Янус-киназы находятся по большей части в цитозоле вблизи к плазматической мембране из-за связывания с рецепторами цитокинов, но TYK2 может быть расположена и в ядре .
Более подробно рассмотрим механизм сигнального пути JAK/STAT и роль янус-киназ в нем (см. рис. 2). Сигнальный путь JAK/STAT состоит из трех компонентов: рецептор, связанный с киназой, янус-киназа и белок STAT, который преобразует передаваемый сигнал и активирует транскрипцию.
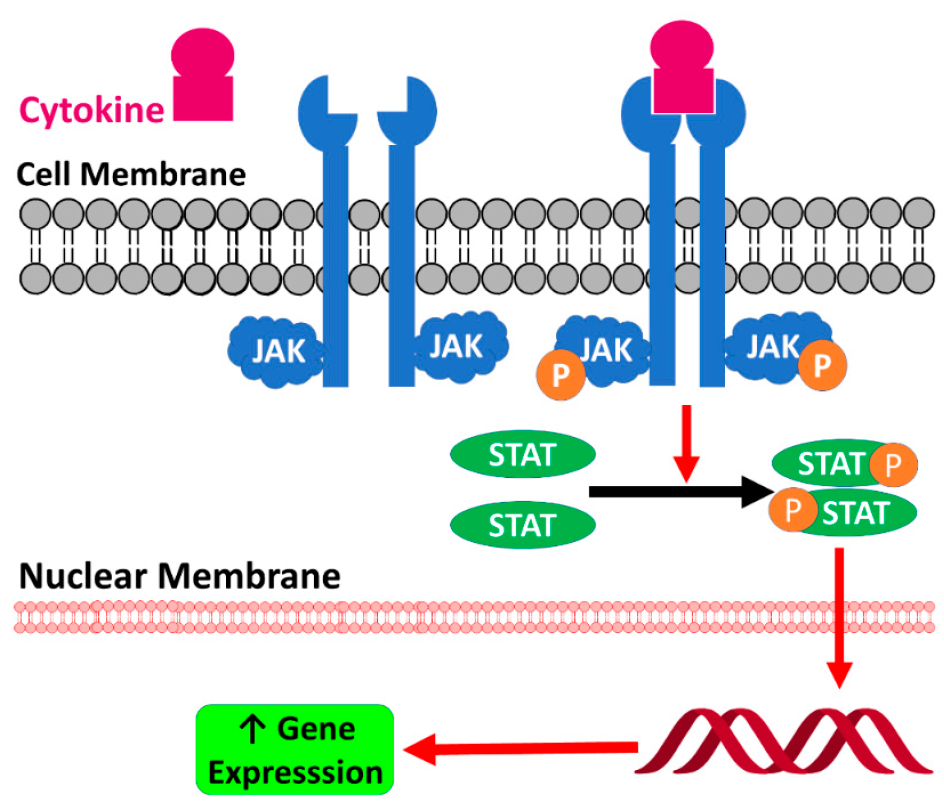
Рисунок 2 - Механизм сигнального пути JAK/STAT
Примечание: по ист. [3]
Возникновение мутаций в янус-киназах приводит к различным злокачественным новообразованиям. Например, мутация JAK2 V617F связана с миелопролиферативными новообразованиями, такими как истинная полицитемия, миелофиброз и другие. Данная мутация находится в псевдокиназном домене и блокирует ингибирование киназного домена. Мутации в 12 экзоне гена JAK2, затрагивающие аминокислотные остатки с 536 по 547, приводят к истинной полицитемии. Мутации JAK1 приводят к острому лимфобластному лейкозу, JAK3 и TYK2 — к острому Т-клеточному лимфобластному лейкозу .
3. Ингибиторы янус-киназ
Низкомолекулярные ингибиторы янус-киназы, так называемые якинибы, являются эффективными и относительно безопасными средствами для лечения многих заболеваний. Якинибы можно разделить на два поколения: первое поколение включает небольшие молекулы, такие как барицитиниб и тофацитиниб, которые действуют как неселективные ингибиторы JAK. Препараты второго поколения, такие как филготиниб и упадацитиниб, обладают селективной ингибирующей активностью в отношении JAK. С другой стороны, якинибы также можно классифицировать в зависимости от способа связывания и типа взаимодействия с аминокислотами в JAK, при этом они делятся на обратимые (конкурентные) и необратимые (ковалентные) ингибиторы (см. рис. 3) .
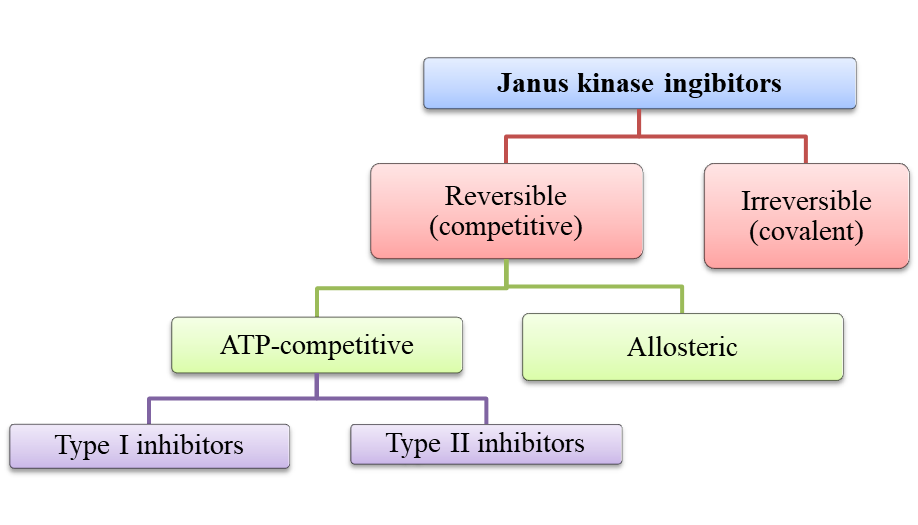
Рисунок 3 - Классификация ингибиторов янус-киназ
Примечание: по ист. [3]
4. Обратимые АТФ-конкурентные ингибиторы янус-киназ I типа
Конкурентные ингибиторы янус-киназ образуют обратимые (нековалентные) взаимодействия с аминокислотами, входящими в состав четырех JAK. Связывающие взаимодействия, образуемые якинибами данного типа, включают водородные связи и гидрофобные взаимодействия. Класс обратимых ингибиторов JAK также можно разделить на два подкласса в зависимости от того с какой именно конформацией JAK происходит взаимодействие. АТФ-конкурентные ингибиторы I типа связываются с АТФ-связывающим сайтом JAK в активной конформации киназного домена, тогда как взаимодействие ингибиторов II типа происходит также с АТФ-связывающим сайтом киназного домена только в неактивной конформации JAK .
Первое поколение якинибов включает соединения, действующие как неселективные ингибиторы (барицитиниб, тофацитиниб и др.).
Барицитиниб (2-[1-этилсульфонил-3-[4-(7H-пирроло[2,3-d]пиримидин-4-ил) пиразол-1-ил] азетидин-3-ил]ацетонитрил) относится к АТФ-конкурентным ингибиторам I типа, используемым для ингибирования JAK1 и JAK2. Он применяется при лечении ревматоидного артрита средней и тяжелой степени у взрослых пациентов, очаговой алопеции , системной красной волчанки . Помимо этого, в 2022 году FDA одобрило барицитиниб для лечения COVID-19 .
Концентрация полумаксимального ингибирования IC50 барицитинибом JAK1 составляет 5,9 нМ, JAK2 — 5,7 нМ. Для TYK2 и JAK3 это значение намного больше: 53 нМ и более 400 нМ соответственно .
Xu J. et al. разработали эффективный пятистадийный метод синтеза барицитиниба с общим выходом 49% (см. рис. 4). Предложенный подход обладает потенциалом для масштабного производства благодаря своей низкой стоимости и простоте технологических требований.
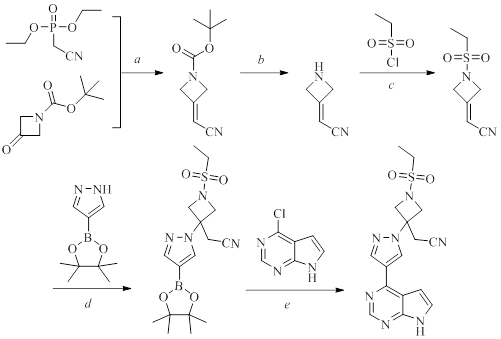
Рисунок 4 - Синтез барицитиниба. Реагенты и условия реакций:
a – NaH/THF, комн. темп.; b – TFA, DCM, комн. темп.; c – DIPEA, CH3CN; d – DBU, IPA, кипячение; e – Pd(PPh3)4, CsF, t-BuOH, вода, толуол, кипячение
Руксолитиниб, также, как и барицитиниб, предотвращает активацию JAK1 и JAK2 со значениями IC50 6,4 и 8,8 нМ соответственно. IC50 для янус-киназы TYK2 равно 19 нМ. Исследования показали, что он оказывает сильное ингибирование активности мутантной JAK2 V617F. Помимо этого, руксолитиниб активен в отношении таких киназ, как c-Src, FAC, RET, TRK-B и LRRK2 (дикий тип) .
При взаимодействии с янус-киназой JAK2 руксолитиниб образует водородные связи между пирролопиримидиновой частью и аминокислотными остатками Glu930 и Leu932 шарнирной области и располагается глубоко внутри АТФ-связывающего кармана. Также руксолитиниб закрепляется в данном положении за счет ван-дер-ваальсовых взаимодействий с окружающими его аминокислотными остатками Leu855, Gly856, Asp994 и Met929. При взаимодействии с янус-киназой JAK1 руксолитиниб связывается с Glu957 и Leu959 .
Руксолитиниб получают пятистадийным синтезом (см. рис. 5). Полученный на стадии d рацемат подвергают хиральному разделению, после обрабатывают трифторуксусной кислотой для снятия защитной группы с R-энантиомера .
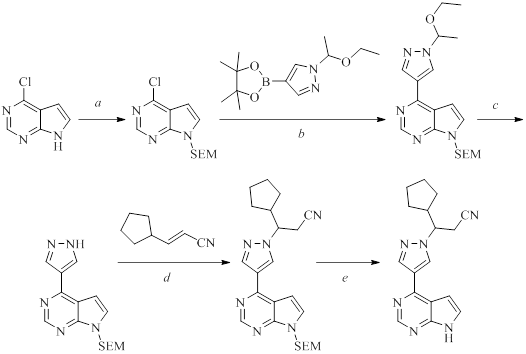
Рисунок 5 - Синтез руксолитиниба. Реагенты и условия реакций:
a – SEM-Cl, DMAC, NaH, 0-5 oC, 35 мин; b – K2CO3, Pd(PPh3)4, n-BuOH/H2O, кипячение, 4 ч; c – HCl, THF, комн. темп.; d – 1. DBU, MeCN, комн. темп.; 2. хиральное разделение; e – TFA, DCM, комн. темп., 6 ч
![Синтез руксолитиниба. Реагенты и условия реакций: a – CuBr, LiBr, THF; b – [{Rh(cod)Cl}2], JoSPOPhos SL-J688-2, PPTS, PhMe, 80 oC, 24 ч; c – 9-BBN, THF, 0 oC до комн. темп. После NaOH, H2O2; d – (COCl)2, DMSO, NEt3, 78 oC потом при комн. темп.; e – I2, NH4OH, THF-H2O, комн. темп.; f – B2pin2, AcOK, [PdCl2(dppf)], DMSO, 90 oC, 24 ч.; g – [PdCl2(PPh3)2], K2CO3, диоксан-вода (2:1), 120 oC, 24 ч](/media/images/2026-02-06/31120d00-f509-40e2-89d8-6d7cf624038a.png)
Рисунок 6 - Синтез руксолитиниба. Реагенты и условия реакций:
a – CuBr, LiBr, THF; b – [{Rh(cod)Cl}2], JoSPOPhos SL-J688-2, PPTS, PhMe, 80 oC, 24 ч; c – 9-BBN, THF, 0 oC до комн. темп. После NaOH, H2O2; d – (COCl)2, DMSO, NEt3, 78 oC потом при комн. темп.; e – I2, NH4OH, THF-H2O, комн. темп.; f – B2pin2, AcOK, [PdCl2(dppf)], DMSO, 90 oC, 24 ч.; g – [PdCl2(PPh3)2], K2CO3, диоксан-вода (2:1), 120 oC, 24 ч
Результаты исследования ингибирующей активности тофацитиниба показали, то он активен в отношении всех четырех янус-киназ. Сильнее всего он ингибирует активность JAK3 – IC50 равно 1,1 нМ. Также тофацитиниб ингибирует JAK1 и JAK2 при значениях IC50 2,9 нМ и 1,2 нМ соответственно, Tyk2 — при 42 нМ. Тофацитиниб взаимодействует с АТФ связывающим центром янус-киназ за счет образования водородных связей, электростатического притяжения и ван-дер-ваальсовых сил .
Синтез тофацитиниба начинают с реакции 4-хлор-7H-пирроло[2,3-d]пиримидина с (3R,4R)-1-бензил-N,4-диметилпиперидин-3-амином (см. рис. 7). Бензильную группу удаляют в ходе Pd/C катализируемого гидрирования, после обработки полученного соединения с 2,5-диоксопирролидинилцианоацетатом получают тофацитиниб, обработка которого лимонной кислотой получают цитрат.
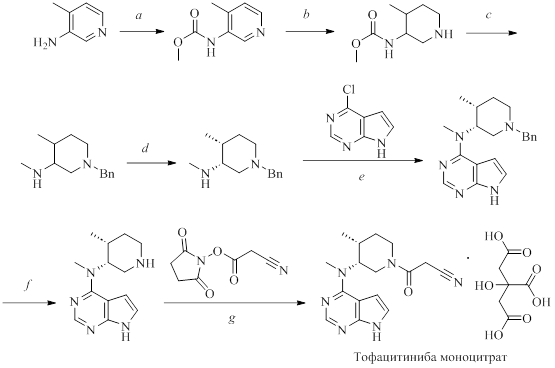
Рисунок 7 - Синтез тофацитиниба. Реагенты и условия реакций:
a – t-BuOK, (MeO)2CO, THF; b – 5% Rh/C, AcOH, H2; c – 1. PhCHO, NaHB(OAc)3, DCM, HOAc; 2. LiAlH4, THF, HCl, IPO; d – L-DTTA; e – Et3N, 100 oC; f – Pd(OH)2/C, H2, EtOH, комн. темп.; g – 1. EtOH, комн. темп., 1 ч; 2. лимонная кислота, ацетон, 40 oC до комн. темп., 2 ч
Takiguchi H. et al. разработали эффективный стереоконтролируемый способ синтеза делгоцитиниба (см. рис. 8).
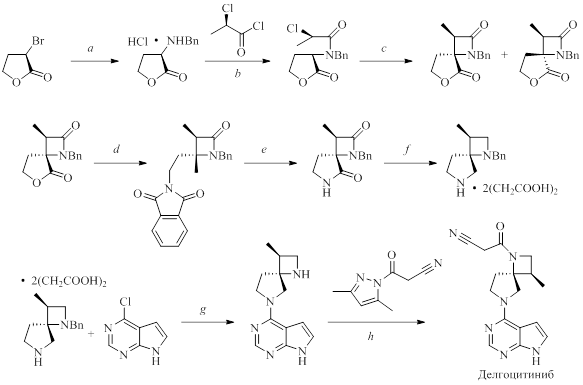
Рисунок 8 - Синтез делгоцитиниба. Реагенты и условия реакций: a – 1. BnNH2, K3PO4, MeCN; 2. 4Н HCl, EtOAc; b – 2,6-лутидин, EtOAc, -10 oC; c – Cs2CO3, DMSO, комн. темп., 4ч; d – 1. KPhth; 2. Etl, DMSO, 100 oC; e – NH(CH2CH2NH2)2, 2-BuOH, 80 oC; f – LiAlH4, TMSCl, толуол, THF, 0-50 oC, 24ч; g – 1. K3PO4, t-BuOH/H2O; 2. H2, Pd/C, AcOH; h – 1. MeCN; 2. Кристаллизация из бутанола
Таблица 1 - Разрешенные к применению АТФ-конкурентные ингибиторы янус-киназ первого поколения I типа
Препарат | Профиль селективности (в порядке убывания) | Год первого одобрения | Основные утверждённые показания (примеры) |
Тофацитиниб | JAK3 > JAK1 > JAK2 (слабо на TYK2) | 2012 (FDA) | Ревматоидный артрит, псориатический артрит, язвенный колит, ювенильный идиопатический артрит, анкилозирующий спондилит. |
Руксолитиниб | JAK1 = JAK2 | 2011 (FDA, США) | Миелопролиферативные заболевания: миелофиброз, истинная полицитемия (при неэффективности гидроксимочевины). |
Баритиицниб | JAK1 = JAK2 > TYK2 > JAK3 | 2017 (ЕС), 2018 (FDA) | Ревматоидный артрит, атопический дерматит (с 12 лет), алопеция ареата, COVID-19 (тяжёлое течение). |
Оклацитиниб | JAK1 > JAK2, JAK3, TYK2. Разработан для ветеринарии. | 2013 (FDA, США) | Атопический дерматит и аллергические дерматозы у собак (первый JAK-ингибитор для животных). |
Пефицитиниб | JAK3 ≥ JAK1 > JAK2 > TYK2 | 2019 (Япония) | Ревматоидный артрит (одобрен в Японии, Южной Корее, Тайване). |
5. Обратимые АТФ-конкурентные ингибиторы янус-киназ I типа второго поколения
Второе поколение якинибов включает селективные ингибиторы (филготиниб, упадацитиниб и др.), которые обладают большей селективностью и меньшими побочными эффектами.
Филготиниб (N-[5-[4-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]фенил]-[1,2,4]триазоло[1,5-а]пиридин-2-ил]циклопропанкарбоксамид) был одобрен в 2020 году Европейским агентством по лекарственным средствам (EMA) для лечения ревматоидного артрита умеренной и тяжелой степени. Филготиниб применяют также для лечения болезни Крона и язвенного колита. Филготиниб встраивается в АТФ связывающий центр JAK1 и образует две водородные связи с Leu932 и одну углерод-водородную связь Lys882. Ингибирующая активность филготиниба по отношению JAK1 наибольшая среди других якинибов и составляет IC50 10 нМ. Кроме этого, филготиниб ингибирует активность hFLT3, hFLT4 и hCSF1R .
Синтез филготиниба — сложный многоэтапный процесс, включающий пять основных стадий , . Изначально эфир 4-(бромметил)фенилбороновой кислоты вводится в реакцию с S,S-диоксидом тиоморфолина. С другой стороны, из 6-бром-2-аминопиридина в результате последовательной обработке этоксикарбонилизотиоцианатом и гидроксиламином, получают 5-бром-[1,2,4]триазоло[1,5-a]пиридин-2-амин. Ацилирование аминогруппы в производном триазола приводит к циклопропанкарбоксамиду, которое в результате реакции кросс-сочетания по Судзуки с полученным на первой стадии эфиром бороновой кислоты превращается в филготиниб (см. рис. 9).
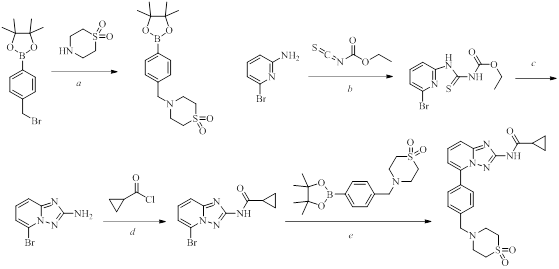
Рисунок 9 - Синтез филготиниба. Реагенты и условия реакций:
a – DIPEA, DCM/MeOH (5:1), комн. темп., 16 ч; b – DCM, 5 oC, до комн. темп.; c – NH2OH, HCl, EtOH/MeOH, DIPEA, комн. темп., 1 ч, после 1-(6-бромопиридин-2-ил)-3-карбоэтокси-тиомочевина, 3 ч, кипячение; d – MeCN, Et3N, 5 oC до комн. темп.; e – 1,4-диоксан/H2O (4:1), K2CO3, PdCl2dppf, 90 oC, 16 ч в атмосфере N2
Аброцитиниб (N-[3-[метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино]циклобутил]пропан-1-сульфонамид) одобрен FDA в январе 2020 года для лечения рефрактерного атопического дерматита средней и тяжелой степени. Он является ингибитором JAK1 при значениях IC50 0,029 нМ и JAK2 при значениях IC50 0,803 нМ. С JAK1 аброцитиниб образует три водородные связи с Glu957, Leu959 и Asn1008, три связи углерод-водород с Leu881 и Leu959 и гидрофобные взаимодействия с другими аминокислотными остатками. С JAK2 аброцитиниб связывается с помощью четырёх водородных связей с Glu930, Leu932, Arg980 и Asn981, одной углерод-водородной связью с Leu932, а также с помощью ван-дер-ваальсовых взаимодействий с Leu855, Val863, Ala880, Val911 и Leu983 .
Vazquez M.L. et al. представили синтез аброцитиниба из циклобутилкарбамата (см. рис. 10).
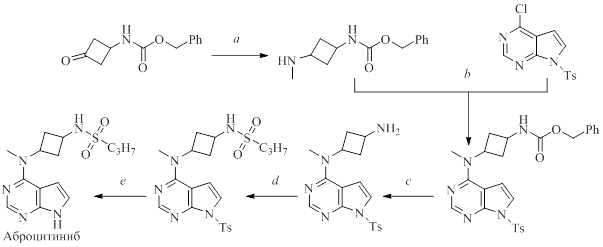
Рисунок 10 - Синтез аброцитиниба. Реагенты и условия реакций:
a – MeNH2, THF, AcOH, комн. темп. потом NaBH4, 0 oC до комн. темп.; b – DIPEA, i-PrOH, 75 oC; c – HBr, AcOH, EtOAc, комн. темп.; d – C3H7SO2Cl, TEA; e – NaOH
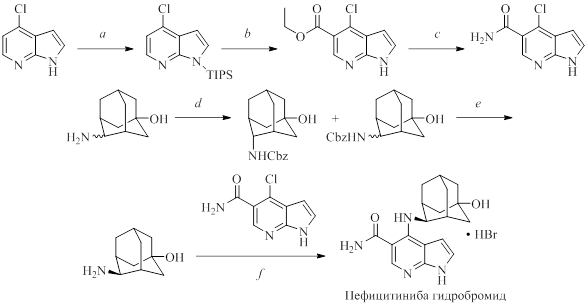
Рисунок 11 - Синтез пефицитиниба. Реагенты и условия реакций:
a – NaH, DMF, 5 oC потом TIPSCl; b – 1. sec-BuLi, THF, -78 oC потом ClCO2Et; 2. TBAF, THF, комн. темп.; c – 1. водн. NaOH, EtOH, 60 oC потом 1М HCl; 2. CDI, DMF, комн. темп. 28% водн. NH4OH; d – CbzCl, водн. NaOH, THF; e – H2, Pd/C, MeOH, комн. темп.; f – 1. TBA, NMP, 200 oC; 2. 48% HBr, EtOAc, кипячение
Синтез упадацитиниба (см. рис. 12) на всех проходит с выходами от хороших до удовлетворительных (52–93%) . После Pd-катализируемого гидрирования полученного соединения, для снятия защитной группы, получают упадацитиниб.
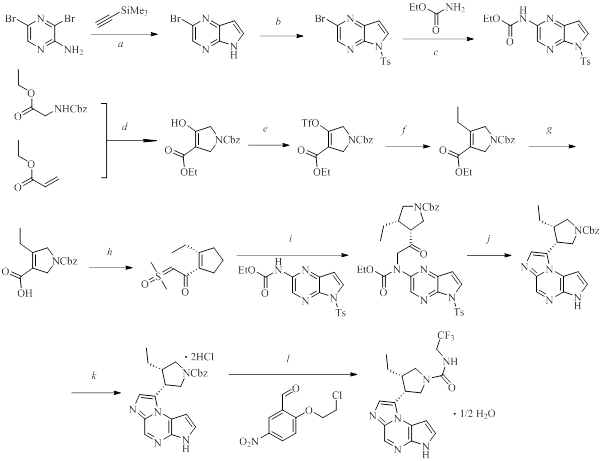
Рисунок 12 - Синтез упадацитиниба. Реагенты и условия реакций:
a – PdCl2(PPh)3, CuI TEA, THF, 0 oC до комн. темп.; b – NaOH, p-TsCl, DMF, 0 oC; c – Pd(OAc)2, ксантфос, K2CO3,PhCH3, 95 oC; d – NaOt-Bu, THF, 0 oC; e – (CF3SO2)2O, DIPEA, i-Pr2O, 0oC; f – EtB(OH)2, PdCl2(dppf), K2CO3, PhCH3, H2O, 85 oC; g – 30% NaOH, THF, 50 oC; h – 1. Ru(OAC)2, (S)сегфос, TEA, H2, MeOH, 80 oC; 2. CDI, THF, 25 oC; 3. (CH3)3SOCl. KOt-Bu, THF, 45 oC до -5 oC; i – 1. LiBr, p-TsOH, THF, 40 oC; 2. Com. LiOt-Bu, DMA, -10 oC; j – пиридин, (CF3COO)2, MeCN, 75 oC, после 20% NaOH, 2-MeTHF, 55 oC; k – 1. Pd(OH)2, EtOH, H2, 50 oC; 2. 35% HCl, EtOH, EtOAc, комн. темп.; l – CDI, THF, 30 oC, 2,2,2-трифторэтан-1-амин, после K2HPO4, THF, H2O, 20% KOH, 25 oC
В таблице 2 приведены одобренные более селективные ингибиторы янус-киназ I типа второго поколения.
Таблица 2 - Разрешенные к применению АТФ-конкурентные ингибиторы янус-киназ I типа второго поколения
Препарат | Профиль селективности (в порядке убывания) | Год первого одобрения | Основные утверждённые показания (на примере FDA/EMA) |
Упадацитиниб | JAK1 > JAK2 > JAK3, TYK2 (селективность к JAK1 ~ 74-кратная относительно JAK2 в клеточных анализах). | 2019 (FDA, США) | Ревматоидный артрит, псориатический артрит, атопический дерматит, аксиальный спондилоартрит, язвенный колит, болезнь Крона. |
Филготиниб | JAK1 > JAK2 (селективность к JAK1 ~ 30-кратная относительно JAK2; практически не активен против JAK3/TYK2). | 2020 (EMA, ЕС; Япония) | Ревматоидный артрит, язвенный колит (в ЕС, Японии). В США не одобрен из-за вопросов безопасности (дозы для мужчин/женщин). |
Аброцитиниб | JAK1 > JAK2 (высокоселективный к JAK1). | 2021 (FDA, США) | Атопический дерматит (с 12 лет). |
Дегоцитиниб | JAK1 > JAK2 = JAK3 > TYK2 | 2020 (Япония) | Атопический дерматит (топическая мазь 0,5%). |
6. Обратимые АТФ-конкурентные ингибиторы янус-киназ II типа
Ингибиторы II типа связываются с тем же АТФ-связывающим карманом, что и ингибиторы I типа, но делают это, когда киназа находится в неактивной (DFG-out) конформации. Это позволяет им занимать дополнительное пространство, образованное поворотом петли DFG, что и обеспечивает потенциально более высокую селективность и способность преодолевать резистентность.
Примерами ингибиторов данного типа являются соединения CHZ868 и BBT594 (рис. 13).
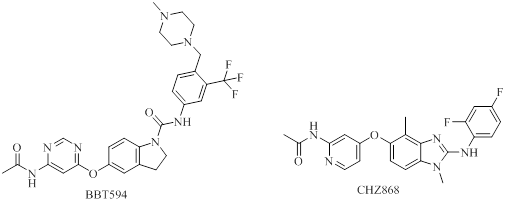
Рисунок 13 - АФТ-конкурентные ингибиторы янус-киназ II типа
![Синтез BBT594. Реагенты и условия реакций: a – 1. NaOH водн., 10 oC до -10 oC, 65 мин; 2. Ацетон, 0 oC, 30 мин; b – 1. Tриэтиламин, C2Cl2, бис(трихлорометил)карбонат, 0 oC, 30 мин; 2. Триэтиламин, C2Cl2, 5-[(6-хлоро-4-пиримидинил)окси]-1Н-индол, 0 oC, 15 мин; c – ацетамид, трис(дибензилиденацетон)дипалладий, (9,9-диметил-9Н-ксантен-4,5-диил)бис[дифенилфосфин], 70 oC, 15 ч](/media/images/2026-02-06/ba1b9532-f06e-4671-8138-4e7b9afae054.png)
Рисунок 14 - Синтез BBT594. Реагенты и условия реакций:
a – 1. NaOH водн., 10 oC до -10 oC, 65 мин; 2. Ацетон, 0 oC, 30 мин; b – 1. Tриэтиламин, C2Cl2, бис(трихлорометил)карбонат, 0 oC, 30 мин; 2. Триэтиламин, C2Cl2, 5-[(6-хлоро-4-пиримидинил)окси]-1Н-индол, 0 oC, 15 мин; c – ацетамид, трис(дибензилиденацетон)дипалладий, (9,9-диметил-9Н-ксантен-4,5-диил)бис[дифенилфосфин], 70 oC, 15 ч
7. Аллостерические ингибиторы янус-киназ
Аллостерические ингибиторы янус-киназ (см. рис. 15) связываются с участком фермента, отличным от АТФ-связывающего кармана. Среди этих ингибиторов в настоящее время одобрен только деукравацитиниб, который действует как селективный аллостерический ингибитор TYK2 .
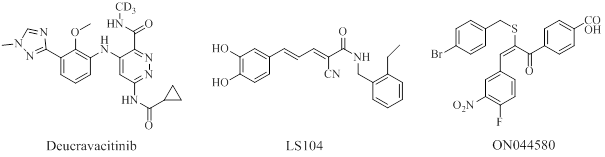
Рисунок 15 - Аллостерические ингибиторы янус-киназ
Кроме того, соединения LS104 и ON044580, проходящие в настоящее время клинические испытания, также являются примерами аллостерических ингибиторов JAK2 , .
Один из способов синтеза деукравацитиниба представлен на рисунке 16 .
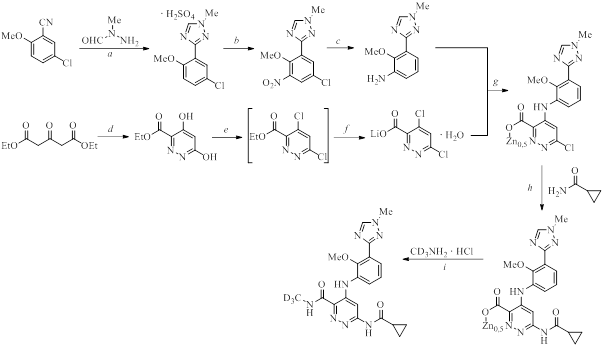
Рисунок 16 - Синтез деукравацитиниба. Реагенты и условия реакций:
a – Kot-Bu, THF; b – HNO3, H2SO4; c – H2, Pd/c, MeOH; d – Ar-SO2N3, n-Bu3P, AcOH, MeCN; e – POCl3, Et3N, MeCN; f – LiBr, DIPEA, aq. MeCN; g – Zn(OAc)2, H2O/IPA; h – Pd(OAc)2, SL-J009, DBU/K2CO3, MeCN/Toluene; i – EDC, HOBt, NMI, NMP/MeCN затем NMP/IPA (2nd drop)
ON044580 ((E)-4-(2-((4-бромбензил)тио)-3-(4-фтор-3-нитрофенил)акрилоил)бензойная кислота), является мощным и неконкурентным с точки зрения АТФ ингибитором киназы JAK2 с IC50 1,23 мкм. ON044580 ингибирует активность киназы JAK2 либо путем связывания с доменом JAK2, связывающим STAT5, либо путем связывания с аллостерическим сайтом .
Препараты ON044580 и LS104 находятся на доклинической стадии разработки.
8. Необратимые (ковалентные) ингибиторы янус-киназ
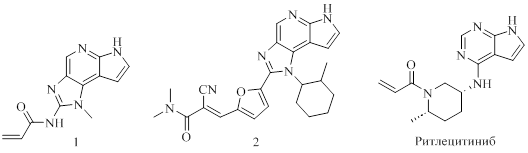
Рисунок 17 - Необратимые (ковалентные) ингибиторы янус-киназ
Усовершенствованный синтетический маршрут (см. рис. 18) позволяет осуществить асимметрический синтез промежуточного этил (2R)-2-((трет-бутоксикарбонил)амино)-5-(((S)-1-фенилэтил)амино)гексаноата, достигая высокого диастереомерного соотношения 86:14. Этил (2R)-2-((трет-бутоксикарбонил)амино)-5-(((S)-1-фенилэтил)амино)гексаноат циклизуется в ключевое промежуточное производное пиперидинового кольца трет-бутил ((3R,6S)-6-метил-2-оксо-1-((S)-1-фенилэтил)пиперидин-3-ил)карбамата под действием уксусной кислоты, что исключает необходимость в дорогостоящих металлических катализаторах. Этот маршрут использует легкодоступные исходные материалы и минимизирует риск загрязнения окружающей среды. Кроме того, он снижает риски для безопасности и необходимость утилизации отходов, подчеркивая экологичность нашего синтетического подхода. Эти достижения не только упрощают синтез ритлецитиниба, но и соответствуют растущему спросу на более экологичные и экономически эффективные синтетические пути в фармацевтической промышленности .
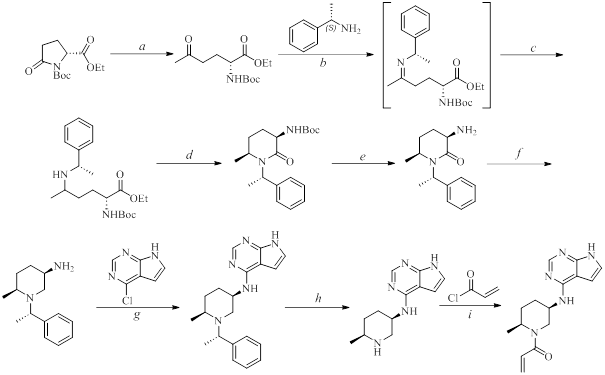
Рисунок 18 - Синтез ритлецитиниба. Реагенты и условия реакций:
a – MeMgCl, THF; b – Ti(OEt)4, THF; c – NaBH4, THF; d – AcOH, toluene; e – HCl, IPA, NaOH aq.; f – NaBH4, TFA, THF; g – 2-Butanol; h – 10%Pd/C, H2, EtOH; i – DIPEA, THF/H2O
9. Безопасность и возможные побочные эффекты якинибов
Ингибиторы янус-киназ второго поколения, такие как филготиниб и упадацитиниб, часто используются для уменьшения воспаления и боли у пациентов с хроническими заболеваниями суставов, при ревматоидном артрите , . Некоторые якинибы применяются для лечения аутоиммунных кожных заболеваний , , например, для лечения узловой эритемы и псориатического артрита.
Клинические испытания якинибов второго поколения показали эффективность в сравнении с традиционными методами лечения, такими как метотрексат , . Они обычно дают более быстрый терапевтический эффект и могут быть использованы в случае, если традиционные препараты не дали нужных результатов. Исследования демонстрируют значительное улучшение симптомов у пациентов после начала терапии ингибиторами янус-киназ и увеличение продолжительности жизни пациентов , . В большинстве исследований отмечается приемлемая степень безопасности, но в связи с подавлением иммунной системы, возможно увеличение риска инфекций, таких как туберкулез , пневмония и другие бактериальные или вирусные заболевания , . Могут наблюдаться гематологические изменения , , например, снижение уровня лейкоцитов и тромбоцитов. Возможны изменения в липидном профиле , что требует мониторинга. В редких случаях могут наблюдаться изменения в показателях функции печени. Иногда возникают проблемы с желудочно-кишечным трактом, включая тошноту и расстройства пищеварения .
10. Заключение
Ингибиторы янус-киназ (JAK-ингибиторы, или «якинибы») утвердились как один из наиболее революционных и динамично развивающихся классов таргетных препаратов, кардинально изменивших подход к терапии широкого спектра тяжёлых иммуноопосредованных, хронических воспалительных и онкогематологических заболеваний. Фармакология прошла путь от первых неселективных препаратов (тофацитиниб, руксолитиниб) к более селективным средствам второго поколения (упадацитиниб, филготиниб, аброцитиниб). Эта эволюция, направленная на повышение избирательности действия в отношении JAK1, позволила сохранить лечебный противовоспалительный эффект, уменьшив при этом влияние на систему кроветворения (связанную с JAK2) и повысив общую безопасность терапии. Современный ассортимент якинибов достаточно разнообразен, так помимо классических обратимых ATP-конкурентных ингибиторов I типа, активно разрабатываются ингибиторы II типа (CHZ868), нацеленные на неактивную конформацию фермента и способные преодолевать резистентность. Прорывным стало появление аллостерических ингибиторов, таких как деукравацитиниб, который, связываясь с псевдокиназным доменом TYK2, открыл принципиально новую мишень и механизм действия. Отдельное направление представляют собой необратимые (ковалентные) ингибиторы (например, ритлецитиниб), формирующие прочную связь с ферментом и обещающие пролонгированный эффект. Такое многообразие создаёт мощный инструментарий для персонализированной терапии.
Несмотря на высокую клиническую эффективность, управление безопасностью ингибиторов янус-киназ остаётся критически важной задачей. К общим для класса рискам, связанным с иммуносупрессией, относятся повышенная восприимчивость к инфекциям (бактериальным, вирусным, включая опоясывающий герпес, грибковым), потенциальные гематологические изменения, а также влияние на липидный профиль и показатели функции печени. Именно развитие селективных и аллостерических препаратов является ключевым путём к смягчению этих побочных эффектов и улучшению долгосрочного баланса пользы и риска.
Перспективы развития якинибов в настоящее время видятся в повышении селективности, преодолении резистентности с помощью инновационных механизмов действия, а также в активном расширении терапевтических показаний на такие заболевания, как алопеция, витилиго и системная красная волчанка. Важными направлениями являются оптимизация лекарственных форм, включая разработку топических препаратов для локальной терапии, и изучение потенциала комбинированной терапии с другими иммуномодуляторами.
Таким образом, ингибиторы янус-киназ представляют собой яркий пример успешной трансляции фундаментальных знаний о внутриклеточной сигнализации в клиническую практику. Постоянно растущее понимание структурной биологии янус-киназ, механизмов развития заболеваний и резистентности открывает путь к созданию лекарственных средств новых поколений. Дальнейшие междисциплинарные исследования в этой области, безусловно, будут способствовать разработке всё более эффективных и безопасных терапевтических стратегий, направленных на улучшение качества жизни миллионов пациентов по всему миру.