FOX-7: AB INITIO STUDY OF THE HIGH ENERGY MATERIALS

Празян Т. Л.
Магистрант, физический факультет, ФГБОУ ВПО КемГУ, Россия, г. Кемерово
FOX-7: ПЕРВОПРИНЦИПНОЕ ИССЛЕДОВАНИЕ ВЫСОКОЭНЕРГЕТИЧЕСКИХ МАТЕРИАЛОВ
Аннотация
Целью настоящей работы является представление данных о структурном и электронном строении FOX-7 в газовой фазе, полученные различными ab initio методами.
Ключевые слова: высокоэнергетические материалы, FOX-7, первопринципные методы.
Prazyan T. L.
Undergraduate student, Department of General Physics, Kemerovo State University, Russian Federation, Kemerovo
FOX-7: AB INITIO STUDY OF THE HIGH ENERGY MATERIALS
Abstract
The aim of this work is to obtain data on the structural and electronic structure of FOX-7 in the gas phase obtained by different ab initio methods.
Keywords: high energy materials, FOX-7, ab initio methods.
Расчеты по оптимизации геометрии, расчету и анализу электронных свойств C2N4O4H4 (далее FOX-7) [1] проводились с использованием программного пакета CRYSTAL09 [2], основанный на методе линейной комбинации атомных орбиталей (ЛКАО). Базисные наборы [3], использовавшиеся при расчетах: C_6-21G*, H_3-1p1G, N_6-31d1G, O_6-31d1. В настоящей работе результаты приведены для FOX-7 в газовой фазе. Как было выяснено ранее, данная модель удовлетворительно описывает свойства изучаемых материалов и дает достаточно точные результаты в сравнении с экспериментальными данными.
На рис.1 (слева) показано молекулярное строение FOX-7. Как можно заметить, рассматриваемая молекула имеет две нитрогруппы и две аминогруппы, атомы кислорода и водорода которых отклонены от основной плоскости молекулы. Справа рис.1 представлена карта распределения электростатического потенциала для молекулы FOX-7. Видно, с аминогрупп вытекает заряд, на это указывают сплошные линии, а не нитрогруппы – натекает. Области отрицательного потенциала обозначены пунктирными линиями, положительного потенциала – сплошными линиями, поверхности нулевого потенциала – штрих-пунктирными линиями.
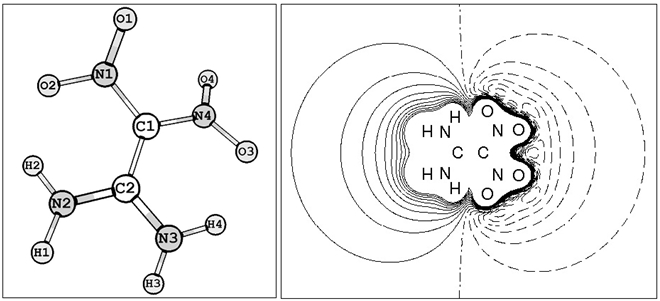
Рис. 1 – Структура молекулы FOX-7 (слева), распределение электростатического потенциала (справа)
Результаты, полученные из первопринципных расчетов в данной работе, представлены в сравнении с экспериментальными данными [4] (таб. 1). Анализируя полученные значения длин связей (в Å), а также сравнив между собой минимумы энергий Etot (в эВ), можно сделать вывод о том, что метод расчета B3LYP, выбранный ранее для изучения высокоэнергетических материалов, таких как PETN, TATB [5] и др., является наиболее приемлемым для описания как структурных и электронных свойств, так и физико-химических [6,7].
Таблица 1 – Минимум энергии и длины связей в молекуле FOX-7
| EXP [4] | B3LYP | HF | LDA | PBE | PWGGA | |
| Etot | -16273.16 | -16192.13 | -16096.68 | -16265.05 | -16277.35 | |
| C1 – C1 | 1.46 | 1.43 | 1.41 | 1.44 | 1.44 | 1.43 |
| C–NO2 | 1.40 | 1.43 | 1.42 | 1.40 | 1.44 | 1.44 |
| C–NH2 | 1.33 | 1.34 | 1.33 | 1.32 | 1.35 | 1.35 |
| N1 – O1 | 1.24 | 1.22 | 1.18 | 1.21 | 1.23 | 1.23 |
| N1 – O2 | 1.25 | 1.25 | 1.21 | 1.26 | 1.27 | 1.27 |
| N2 – H1 | 1.01 | 1.01 | 0.99 | 1.02 | 1.01 | 1.01 |
| N2 – H2 | 1.01 | 1.01 | 0.99 | 1.08 | 1.04 | 1.03 |
Также из первых принципов получен частотный спектр (рис. 2). При частоте 1296 см-1 и интенсивностью 556 км/моль колеблются атомы азота N1 и N4, а также атомы водорода H1 и H3. При частоте 1638 см-1 и интенсивностью 1028 км/моль – атомы углерода C1 и C2, атомы водорода H1 – H4. При частотах 3436, 3459 и 3705 см-1 и интенсивностями 210, 104 и 131 км/моль, соответственно, колеблются атомы водорода H1 – H4.
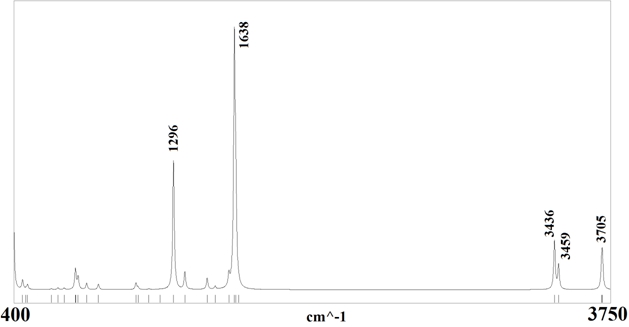
Рис. 2 – Частотный спектр молекулы FOX-7
Литература- Appalakondaiah S. Structural, vibrational, and quasiparticle band structure of 1, 1 -diamino-2, 2 - dinitroethelene from ab-initio calculations / S. Appalakondaiah, G. Vaiheeswaran // The J. Of Chem. Phisics. – 2014. - № 140 (1). – P. 1-16.
- Dovesi R. CRYSTAL09 User’s Manual / R. Dovesi, V.R. Saunders, C. Roetti [et al.]. - Torino: University of Torino, 2010.
- CRYSTAL Basis Sets Library [Электронный ресурс] URL: http://www.crystal.unito.it/Basis_Sets/Ptable.html (дата обращения 24.02.2015).
- Bemm U., Ӧstmark H. Acta Crystallogr., 1998. P. 1997.
- Valenzano L. Accurate prediction of second-order elastic constants ftom first principles: PETN and TATB / L.Valenzano, W. J. Slough, W. F. Perger // AIP Conf. Proc. -2012. - № 1426. – P. 1191-1194.
- Празян Т. Л. Исследование физико-химических свойств ряда взрывчатых веществ методами компьютерного моделирования / Т. Л. Празян, Ю. Н. Журавлев // Вестник КемГУ. – 2014. – № 4 (60). – Т. 2. – C. 137-144.
- Appalakondaiah S. Structural, vibrational, and quasiparticle band structure of 1, 1 -diamino-2, 2 - dinitroethelene from ab-initio calculations / S. Appalakondaiah, G. Vaiheeswaran // The J. Of Chem. Phisics. – 2014. - № 140 (1). – P. 1-16.
- Dovesi R. CRYSTAL09 User’s Manual / R. Dovesi, V.R. Saunders, C. Roetti [et al.]. - Torino: University of Torino, 2010.
- CRYSTAL Basis Sets Library [Jelektronnyj resurs] URL: http://www.crystal.unito.it/Basis_Sets/Ptable.html (data obrashhenija 24.02.2015).
- Bemm U., Ӧstmark H. Acta Crystallogr., 1998. P. 1997.
- Valenzano L. Accurate prediction of second-order elastic constants ftom first principles: PETN and TATB / L.Valenzano, W. J. Slough, W. F. Perger // AIP Conf. Proc. -2012. - № 1426. – P. 1191-1194.
- Prazyan T. L. Issledovanie fiziko-himicheskih svojstv rjada vzryvchatyh veshhestv metodami komp'juternogo modelirovanija / T. L. Prazyan, Ju. N. Zhuravlev // Vestnik KemGU. – 2014. – № 4 (60). – T. 2. – S. 137-144.