DEVELOPMENT AND VALIDATION OF A TEST SYSTEM FOR ANALYSING THE POLYMORPHISM OF THE COMPLETE MITOCHONDRIAL GENOME OF PIGS
DEVELOPMENT AND VALIDATION OF A TEST SYSTEM FOR ANALYSING THE POLYMORPHISM OF THE COMPLETE MITOCHONDRIAL GENOME OF PIGS
Abstract
In this study, a primer test system was developed and PCR conditions were optimized for the amplification of complete sequences of the mitochondrial genome of pigs. The validation of the developed test system was carried out on samples of four transboundary pig breeds and wild boar from two regions of the country. As a result, nucleotide sequences of complete mitochondrial genomes of swine (n=17) and wild boar (n=3) samples were obtained with a length of 16611 bp. 16 haplotypes were identified in the studied sample of animals, 13 of which were found in domestic pigs and 3 in wild boar. No common haplotypes were detected among domestic pigs and wild boar. Based on the obtained complete MT-DNA sequences, the calculation of genetic diversity parameters was performed, which revealed that wild boars outperformed pig breeds both in terms of the average number of nucleotide differences and nucleotide diversity values: k=106.00 and π=0.00638. Phylogenetic analysis showed that, despite the small sample of animals studied, all five known haplogroups of the genus Sus srofa A, B, C, D, and E were determined, which indicates the universality of the developed test system.
1. Введение
Домашняя свинья (Sus scrofa domestic) является наиболее распространенным и экономически значимым видом сельскохозяйственных животных в России. По данным статистики Росстат производство свинины в России растет год от года. В 2022 году производство свинины составило 4,52 млн т в убойной массе, уступив лишь отрасли птицеводства (5,3 млн т). Поэтому, основная доля обеспечения населения страны мясом и мясопродуктами приходится на свиноводство.
Племенная база свиноводства России на начало 2022 г. представлена 7 породами свиней с общей численностью основных и проверяемых свиноматок 122280 голов. При этом основной разводимой породой является крупная белая, удельный вес которой составляет 56,9%, далее следуют: йоркшир – 18,52%, ландрас – 18,18%, дюрок – 5,83%, на остальные породы – приходится 0,56% .
Для эффективного ведения данной отрасли необходимо правильное и полное использование ее производственного потенциала, что становится невозможным без использования данных о генетических механизмах, происходящих в породах свиней, разводимых на территории России.
Всемирная Продовольственная и сельскохозяйственная организация ФАО считает основными трудностями в рациональном использовании пород сельскохозяйственных животных отсутствие полноценной информации об их генетической структуре . В связи с этим изучение и сохранение биоразнообразия отечественного генофонда свиней, которые представляют значимый экономический интерес для страны, является актуальной задачей в современном мире.
Для характеристики генетического разнообразия домашних животных и их диких сородичей, в частности домашних свиней и диких кабанов, на сегодняшний день используется множество маркеров, таких как микросателлиты (STR) , однонуклеотидные полиморфизмы ядерной ДНК (SNP) , полиморфизмы митохондриальной ДНК .
В то время как в России повсеместно проводятся работы по изучению генетического разнообразия домашних свиней, основанные на исследовании полиморфизмов ядерной ДНК (STR- и SNP-маркеры), митохондриальный геном остается без должного внимания. На сегодняшний день митохондриальная ДНК российских пород свиней исследуется только по единичным маркерам, таким как D-петля , ген COX2 . Однако, остальные гены остаются малоизученными. При этом, мтДНК является единичным маркером, так как ведет себя как один нерекомбинантный локус. Исследование полной последовательности митохондриального генома позволит всесторонне изучить роль мтДНК в жизнедеятельности свиней. В связи с этим, актуальной задачей становится разработка тест-системы анализа полиморфизма полных митохондриальных геномов свиней (Sus Scrofa).
В связи с вышеизложенным целью данной работы являлась разработка и валидация тест-системы анализа полиморфизма полных нуклеотидных последовательностей митохондриальных геномов свиней (Sus Scrofa).
2. Методы и принципы исследования

Рисунок 1 - Электрофореграмма проверки целостности, выделенной ДНК
В соответствии с референсной последовательностью полного митохондриального генома свиней, представленной в базе Национального центра биотехнологической информации NCBI (GenBank: NC_000845.1) с помощью онлайн-ресурса Basic Local Alignment Search Tool (BLAST) были подобраны праймеры для амплификации четырех перекрывающихся фрагментов мтДНК длиной от 4576 до 4857 п.о. (табл. 1).
Таблица 1 - Последовательности праймеров, подобранных для амплификации последовательностей полного митохондриального генома свиней
№ | Последовательность | Позиция (длина) | t/GC % | ПЦР (п.н.) |
1 | F- 5`-TCGGACAACCAGCTATCACCA-3` | 13976-13996 (21) | 61/52 | 4701 |
R- 5`-CATACACCGCCTACCAACATA-3` | 22084-2064 (21) | 60/48 | ||
2 | F- 5`-TATTAGCGGTACGAGTCAGTTA-3` | 9846-9868 (24) | 62/42 | 4714 |
R- 5`-CTTGATTGAATAAGGCCATGAAGC-3` | 14560-14537 (24) | 62/42 | ||
3 | F- 5`-GTCAAATCCACATTCATATGGGC-3` | 5836-5858 (22) | 61/43 | 4576 |
R- 5`-CTAAGGACTGCAGGACTTATC-3` | 10412-10392 (21) | 60/48 | ||
4 | F- 5`-TGGAGTTGGTTGTGGTATTGTATT-3` | 1458-1481 (24) | 60/38 | 4857 |
R- 5`-ATCCAAGCACTATCCATCACCAT-3` | 6337-6315 (23) | 61/43 |
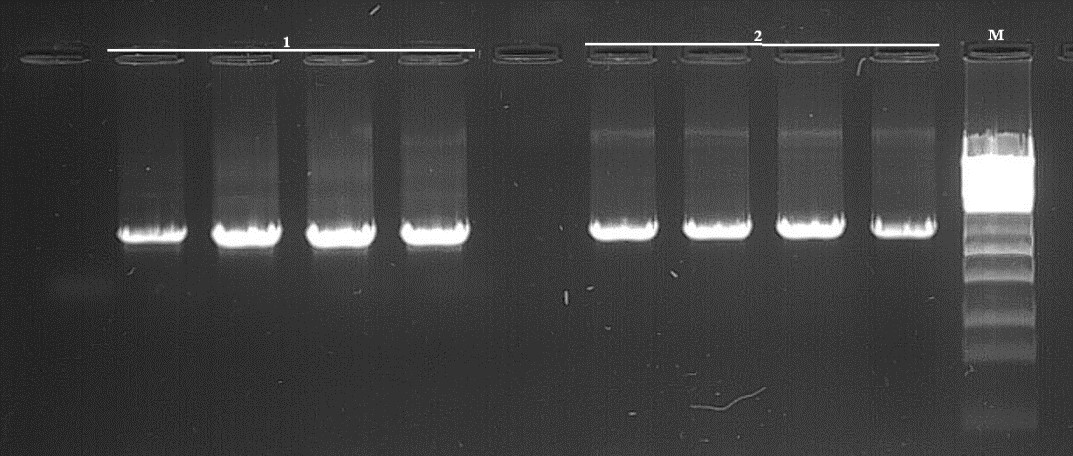
Рисунок 2 - Электрофореграмма 1% агарозного геля
Примечание: длина ампликона составляет от 4578 до 4857 п.о.
Прочтение нуклеотидных последовательностей, полученных ампликонов мтДНК, проводили методом секвенирования нового поколения NGS (next generation sequencing) на приборе miSeq (Illumina, США). Создание библиотек для дальнейшего секвенирования полученных продуктов ПЦР проводили с использованием набора NEBNext Ultra II DNA Library Prep Kit for Illumina согласно протоколу производителя. Для секвенирования подготовленных библиотек использовались наборы реагентов MiSeq Reagent Micro Kit, v2 (300 cycles) для прямого и обратного прочтения (Illumina, США).
Биоинформатическая обработка полученных ридов проводилась с использованием следующих программ: FastQC 0.11.9 – проверка качества «сырых» данных; Trimmomatic 0.3.9 – тримминг ридов; bowtie2 v2.4.4 – картирование ридов. Построение филогенетического дерева методом ML (максимального правдоподобия) выполнялось в программе MEGA Х .
С использованием программы DnaSP 6.12.01 , был проведен расчет параметров генетического разнообразия: число полиморфных сайтов (S), среднее число нуклеотидных различий (K), количество гаплотипов (H), гаплотипическое разнообразие (Hd), нуклеотидное разнообразие (π).
Для определения принадлежности исследуемых образцов свиней и кабана к известным гаплогруппам, из базы NCBI были отобраны последовательности мтДНК свиней, относящиеся к гаплогруппам А, В, С, D и E (табл. 2).
Таблица 2 - Номера последовательностей известных гаплогрупп свиней
№ п/п | Гаплогруппы | № NCBI |
1 | A | KT279758.1 |
2 | B | KT261429.1 |
3 | C | KT279759.1 |
4 | D | KT279760.1 |
5 | E | KT261430.1 |
3. Основные результаты
Нами были получены нуклеотидные последовательности полных митохондриальных геномов образцов свиней и дикого кабана (n=20) длиною 16611 п.н. Для расчетов параметров генетического разнообразия, исследуемые образцы были распределены на две группы: домашние свиньи и дикие кабаны (табл. 3). Наибольшее среднее число нуклеотидных различий было обнаружено у диких кабанов по сравнению с домашними свиньями: k=106,00 и k=90,985, соответственно. Кроме того, дикие кабаны характеризовались большим нуклеотидным разнообразием (π=0,00638) в сравнении с домашними свиньями (π=0,00548). При этом количество полиморфных сайтов было больше у домашних свиней по сравнению с дикими кабанами: S=237 и S=159, соответственно.
Таблица 3 - Показатели генетического разнообразия домашних свиней и диких кабанов
Группа | n | S | H | Нd | k | π |
Домашняя свинья | 17 | 237 | 13 | 0,971± 0,028 | 90,985 | 0,00548±0,00051 |
Дикий кабан | 3 | 159 | 3 | 1,000±0,272 | 106,00 | 0,00638±0,00266 |
Всего | 20 | 244 | 16 | 0,979±0,021 | 89,753 | 0,00541±0,00040 |
Примечание: n – число образцов, S – число полиморфных сайтов, K – среднее число нуклеотидных различий, H – количество гаплотипов, Hd – гаплотипическое разнообразие, π – нуклеотидное разнообразие
Всего было определено 16 гаплотипов, в том числе 13 у домашних свиней и 3 у диких кабанов. Все выявленные гаплотипы домашних свиней отличались от гаплотипов, обнаруженных у кабанов.
В породах свиней крупная белая и йоркшир, а также в группе диких кабанов были выявлены породоспецифичные гаплотипы. Один гаплотип (Hap-4) был общим для пород ландрас и дюрок (табл. 4).
Таблица 4 - Распределение гаплотипов по породам
Гаплотип | Породы | Гаплотип | Породы |
Hap-1 | Крупная белая | Hap-9 | Крупная белая |
Hap-2 | Йоркшир | Hap-10 | Крупная белая |
Hap-3 | Йоркшир | Hap-11 | Йоркширская |
Hap-4 | Ландрас/Дюрок | Hap-12 | Дюрок |
Hap-5 | Крупная белая | Hap-13 | Йоркшир |
Hap-6 | Крупная белая | Hap-14 | Дикий кабан |
Hap-7 | Крупная белая | Hap-15 | Дикий кабан |
Hap-8 | Крупная белая | Hap-16 | Дикий кабан |
Анализ филогенетического дерева (рис. 3), построенного методом ML (максимального правдоподобия) выявил, что гаплотипы домашних свиней Hap-5 и Hap-6 были соотнесены с представителями гаплогруппы A. Гаплотипы Hap-1, Hap-3, Hap-10 домашних свиней и гаплотип Hap-14 дикого кабана были определены с гаплогруппой B. С гаплогруппой C кластеризовались гаплотипы Hap-2, Hap-13, Hap-12 домашних свиней и гаплотип Hap-15 дикого кабана, а гаплотипы Hap-4, Hap-7, Hap-11 домашних свиней и гаплотип Hap-16 дикого кабана были распределены с гаплогруппой D, в то время как гаплотипы Hap-8 и Hap-9 были определены с представителями гаплогруппы E.
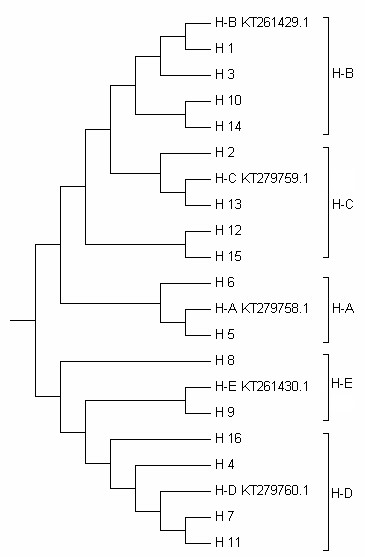
Рисунок 3 - Филогенетическое дерево, построенное методом максимального правдоподобия
4. Заключение
Таким образом, разработанная нами тест-система, позволила получить полные митохондриальные геномы домашних свиней и дикого кабана. В результате нами были получены 20 последовательностей мтДНК длиной 16613 п.н., на основании которых, была дана характеристика материнской изменчивости и определена гаплогрупповая принадлежность исследуемых образцов к известным, на сегодняшний день, гаплогруппам рода Sus sсrofa A, B, C, D, и E.
Полученные в данной работе результаты исследований открывают перспективы дальнейшего проведения обширного секвенирования различных пород свиней и популяций кабана, обитающих на территории России, что позволит получить детальную информацию не только о состоянии генетического разнообразия, но и о древних эволюционных событиях и демографической истории представителей вида Sus scrofa.