ГЕТЕРОАНАЛОГИ ПРОСТАГЛАНДИНЭНДОПЕРОКСИДА НА ОСНОВЕ СТЕРЕОИЗОМЕРНЫХ 3,4,5,6-ТЕТРАХЛОРТРИЦИКЛО[6.2.1.02,7]-УНДЕКА-3,5-ДИЕН-9,10 ДИКАРБОНОВЫХ КИСЛОТ

Исмаилов С.А.
Доктор химических наук, Институт нефтехимических процессов Академии наук Азербайджана
ГЕТЕРОАНАЛОГИ ПРОСТАГЛАНДИНЭНДОПЕРОКСИДА НА ОСНОВЕ СТЕРЕОИЗОМЕРНЫХ 3,4,5,6-ТЕТРАХЛОРТРИЦИКЛО[6.2.1.02,7]-УНДЕКА-3,5-ДИЕН-9,10 ДИКАРБОНОВЫХ КИСЛОТ
Аннотация
На базе аддуктов реакции 5,5-диметокситетрахлорциклопентадиена с стереоизомерными эндо- и экзо-бицикло[2.2.1]гепт-5-ен-2,3-цис-(транс-) кислотами синтезированы гетероаналоги простагландинэндопероксида
Ключевые слова: простагландинэндопероксид, простациклин, тромбоксан, гетеропростаноиды, дициклогексилкарбодиимид.
Ismailov S.A.
Doctor of Chemical Sciences. Institute of Petrochemical Processes of Azerbaijan Academy of Scienes
HETEROANALOGS OF PROSTAGLANDINENDOPEROXIDE ON THE BASIS OF STEREOISOMER 3,4,5,6-TETRACHLORTRICYCLO[6.2.1.02,7]-UNDEKA-3,5-DIEN-9,10-DICARBOXYLIC ACIDS
Abstract
Heteroanalogs of prostaglandinendoperoxide have been synthesized on the basis of adducts of reaction of 5,5-dimethoxytetrachlorcyclopentadiene with stereoisomer endo- and exo-bicyclo[2.2.1]hept-5-en-2,3-cis-(trans-)acids.
Keywords: prostaglandinendoperoxide, prostacyclin, thromboxane, heteroprostanoides, dicyclohexylcarbodiimide.
В отличие от других классов биоактивных соединений работы по модифицированию структур простаноидов были начаты практически сразу после открытия и установления структуры. По интенсивности проводимых работ и практической значимости полученных результатов область модифицированных простаноидов сравнимы лишь с обширными достижениями β-лактамных антибиотиков [1,2].
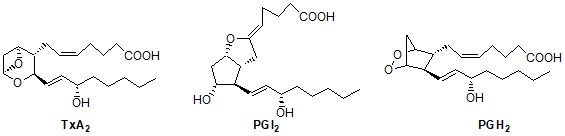
Как известно, природные простаноиды проявляют биологические эффекты в крайне низких нанограммовых дозах, однако отличаются химической и метаболической неустойчивостью и нежелательной широтой спектра физиологического действия [3]. Целенаправленной модификацией простагландиновой молекулы удалось синтезировать более устойчивые, селективно и пролонгировано действующие аналоги [4]. Из их числа в плане создания антитромбозных, сердечно-сосудистых средств перспективны модификаты простагландина I2 и в ряде случаев тромбоксана А2 и простагландинэндопероксида [5-7]. Если ТхА2 стимулирует агрегацию тромбоцитов и обладает вазоконстрикторным действием, то эффект PGI2 прямо противоположный. Оба эти соединения in vivo образуются из единого предшественника PGH2. Для поддержания нормального состояния организма важное значение имеет оптимальный баланс TxA2 и PGI2. В патологических состояниях равновесие TxA2 – PGI2 нарушено, имеет место избыточный биосинтез в организме TxA2. Терапия болезней, связанных с тромбобразованием заключается в блокировании путей биосинтеза TxA2 ксенобиотиками, в число которых входят и некоторые аналоги PGH2, способные уменьшать уровень простагландинэндопероксида [2-10]. В частности, показано, что заменой эндопероксидного мостика на этановый в PGH2, а также модифицирование боковых цепей гетероатомом (N, S, O) можно усилить биологические свойства, в том числе, антиагрегационную активность [11-16].
Исходя из этих соображений, в данной работе описываем синтезы стереоизомерных гетеропростаноидов I, VII и XII, ретроспективные схемы которых представлены ниже:
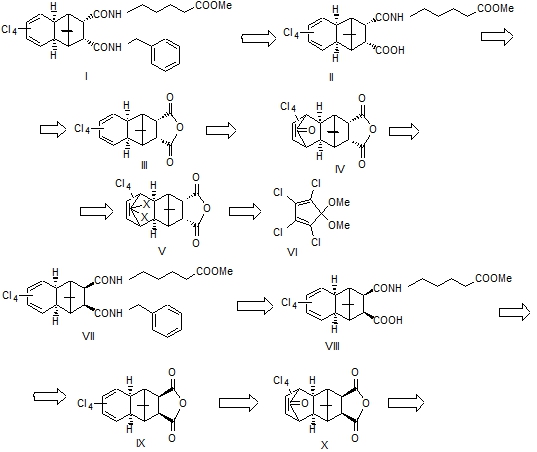
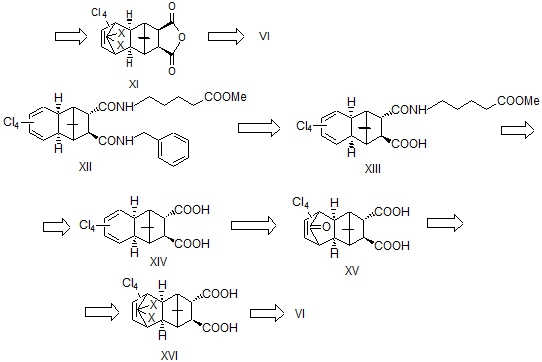
Ранее нами впервые были проведены [17] реакции 5,5-диметокситетрахлорцик-лопентадиена VI c ангидридами эндо- и экзо-бицикло[2.2.1]гепт-5-ен-2,3-дикар-боновой кислоты (БГДК), а также транс-БГДК. Была изучена стереохимия полученных соответствующих аддуктов III, IX, XIV методами ЯМР ¹Н и рентгеноструктурного анализа [18,19]. В данной работе предприняли осуществить их превращение через некоторые манипуляции в целевые простаноиды I, VII, XII – аналоги PGH2 c глубокими структурными изменениями.
Поскольку стандартная последовательность построения боковых цепей простаноидов не приемлема для субстратов III, IX, XIV (тетрахлордиеновая система довольно лабильна при воздействии сильных восстановителей и щелочных агентов) мы построили боковые цепи с амидной связью.
Установили, что экзо-эндо-III и экзо-экзо-ангидриды IX в среде триэтиламина при комнатной температуре легко взаимодействуют с гидрохлоридом метилового эфира δ-аминовалериановой кислоты, давая с количественными выходами соответствующие стереоизомерные амидокислоты II и VII. Следует отметить, что в каждом отдельном случае реакция идет стереоспецифично с образованием одного индивидуального аддукта. Для построения второй боковой цепи через амидную связь в качестве активатора группы СООН использовали дициклогексилкарбодиимид (ДСС) и с высокими выходами получили эндо-цис-I и экзо-цис-простаноиды VII изо-строения.
Для получения транс-эпимера соединений I и VII нами использована транс-эпимерная кислота XIV. При этом следовало ожидать определенной хемоселективности при амидировании эндо- и экзо-карбоксильных групп кислоты XIV и выйти таким образом к одному из эпимеров. Однако при экспериментальной проверке была получена неразделимая на ТСХ смесь региоизомеров XIII и XIIIa в равных соотношениях (по данным ВЭЖХ). В спектре ЯМР ¹³С этой смеси соединений также наблюдается удвоение сигналов. Далее, проведением последующей реакции амидирования соединений XIII и XIIIa с бензиламином в присутствии ДДС получили соответствующую смесь региоизомеров XII и XIIa в соотношении 1:1.5, которые были разделены методом ВЭЖХ.
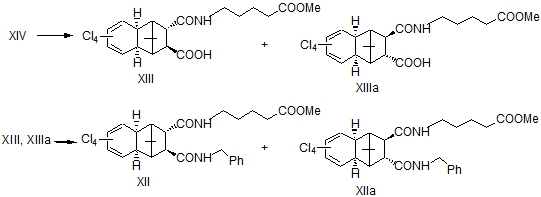
Таким образом, исходя из стереоизомерных ангидридов экзо-эндо-III и экзо-экзо-1,2-дикарбоновых кислот IX, а также транс-кислоты XIV были синтезированы α,ω-бисамидные гетеропростаноиды изо- и нормального строения.
Экспериментальная часть
ИК спектры сняты на спектрофотометре Specord M-80 d в жидкой пленке и в суспензии в вазелиновом масле. Спектры ЯМР ¹Н и ¹³С записаны на приборе Bruker AM-300 с рабочей частотой 300 и 75 МГц соответственно. Внутренний стандарт – ТМС, растворитель CDCl3. Анализ и препаративное разделение соединений методом ВЭЖХ проводили на жидкостном хроматографе Du Pont – 800 с использованием колонок Zorbax Sil 4.6x250 мм (анализ) и 21.2х250 мм (разделение), размер заполненного сорбента 5 мк, детектор – рефрактометр R-401 (фирма Waters). Для качественного анализа ТСХ использовали пластинки Silufol UV-254 с обнаруживанием веществ с помощью УФ-облучения (λ 254) и иодом. Разделение продуктов реакции проводили методом колоночной хроматографии на силикагеле L 40/100, L 100/160 и L 100/250 (Chemapol, ЧССР), элюент – гексан-этилацетат.
9-(4-Метоксикарбонилбутиламид)-3,4,5,6-тетрахлортрицикло[6.2.1.02,7]унде-ка-3,5-диен-эндо-цис-9,10-дикарбоновой кислоты II.
К смеси 1.0 г ангидрида III, 1.0 г триэтиламина в 15 мл ацетона по каплям прибавляли 0.5 г гидрохлорида метилового эфира δ-аминовалериановой кислоты в 10 мл ацетона. Реакция быстро совершается за 10 мин. Упарили ацетон, к остатку прибавляли воду и подкисляли разбавленной HCl. При этом выпавшие кристаллы отфильтровывали, промыли ацетоном и сушили. Получено 1.3 г соединения II с т.пл. 175-177ºС. ИК спектр(см-1): 1760, 1770 (C=O), 1610 (C=C). Спектр ЯМР ¹Н (ДМФА-d7, δ, м.д.): 7.50 уш.с. (2Н, NH и COOH), 3.75 д.д. (2Н, С9Н, С10Н, J¹ 15.4 Гц, J² 5.4 Гц), 3.60 с (3Н, СООСН3), 3.25 м (2Н), 2.98 м (4Н), 2.25 м (2Н), 1.65 д.д. (2Н,С11Н2, J¹=J²=12.5 Гц), 1.50 м (2Н). Спектр ЯМР ¹³С (ДМФА-d7, м.д.): 174.51 с (С=О), 173.23 с (С=О), 172.13 с (С=О), 133.21 с (-CCl=), 132.05 c (-CCl=), 123.60 c (-CCl=), 122.77 c (-CCl=), 52.01 k (OCH3), 49.90 д (СН), 49.30 д (СН), 47.79 д (СН), 47.60 д (СН), 46.89 д (СН), 46.16 д (СН), 39.23 т (С11Н2), 36.02 т (СН2), 35.96 т (СН2), 29.80 т (СН2), 22.90 т (СН2).
Аналогично из экзо-ангидрида IX получили 9-(4-Метоксикарбонилбутил-амид)-3,4,5,6-тетрахлортрицикло[6.2.1.02,7]ундека-3,5-диен-экзо-цис-9,10-дикарбоновой кис-лоты VIII с выходом 86%. Т.пл. 175-176 ºС. ИК спектр (см־¹): 1760, 1770 (С=О), 1610 (С=С). Спектр ЯМР ¹Н (ДМФА-d7 , δ, м.д.): 8.67 уш.с (2Н, NH и COOH), 3.65 с (3Н, СООСН3), 3.20 м (2Н), 3.00 м (4Н), 2.95 м (2Н), 2.75 м (2Н), 2.41 с (2Н), 2.35 т (2Н, J 7.50 Гц), 1.60 д.д. (2Н, С11Н2, J¹=J²=12.5 Гц), 1.58 м (2Н). Спектр ЯМР ¹³С (ДМФА-d7, δ, м.д.): 174.05 с (С=О), 173.86 с (С=О), 172.39 с (С=О), 132.75 с (-ССl=), 132.32 c (-CCl=), 123.47 c (-CCl=), 123.31 c (-CCl=), 52.08 k (OCH3), 51.50 д (СН), 51.29 д (СН), 51.23 д (СН), 50.18 т (СН2), 47.96 д (СН), 39.17 т (СН2), 33.73 т (СН2), 32.92 т (СН2), 29.34 т (СН2), 22.82 т (СН2).
9-Бензиламид-10-(4-метоксикарбонилбутиламид)-3,4,5,6-тетрахлортри- цикло[6.2.-1.02,7]ундека-3,5-диен-эндо-цис-9,10-дикарбоновой кислоты I.
К перемешиваемому раствору 0.4 г амида II в 10 мл абс. ТГФ прибавляли по каплям (20 ºС) раствор 0.2 г ДДС в 10 мл ТГФ. Через 30 мин. образуется белый осадок. Затем добавляли 0.15 г бензиламина в 10 мл абс. ТГФ. Перемешивание продолжали 4 ч. Осадок отфильтровывали, промывали ТГФ и фильтрат подвергали упариванию. Остаток хроматографировали на силикагеле (элюент – хлористый метилен-метанол, 8:1). Получено 400 мг продукта I. Т.пл. 105-107 ºС. ИК спектр (см־¹): 1760, 1775 (С=О), 1608 (С=С). Спектр ЯМР ¹Н (ДМФА-d7, δ, м.д.): 8.25 уш.с (2Н, 2NH), 7.30 м (5Н, Ph), 4.35 д.д.д.(2Н, С9Н, С10Н, J¹=J²=5.9 Гц, J³=15.2 Гц), 3.95 с (2Н, СН2, бензила), 3.63 с (3Н, СООСН3), 3.12 м (4Н, 4СН), 2.90 м (2Н, СН2), 2.35 т (2Н, J 7.52 Гц), 1.70 м (2Н, СН2), 1.65 д.д. (2Н, С11Н2, J¹=J²=12.1 Гц), 1.58 м (4Н, 2СН2). Спектр ЯМР ¹³С (ДМФА-d7, δ, м.д.): 174.05 с (С=О), 171.54 с (С=О), 140.63 с (С¹ бензила), 133.58 с (-ССl=), 128.79 д (СН бензила), 128.54 д (СН бензила), 127.27 д (СН бензила), 123.07 с (-ССl=), 123.02 с (-ССl=), 51.47 к (ОСН3), 49.94 д (СН), 47.24 т (СН2 бензила), 47.19 д (СН), 43.18 д (СН), 39.02 т (СН2), 34.66 т (СН2), 29.86 т (СН2), 25.42 т (СН2), 22.87 т (СН2).
Аналогичо из амида VIII синтезировали 9-Бензиламид-10-(4-метоксикарбо-нилбутиламид)-3,4,5,6-тетрахлортрицикло[6.2.1.02,7]ундека-3,5-диен-экзо-цис-9,10-дикарбоновой кислоты VII с выходом 65%. Т.пл. 165-167 ºС. ИК спектр (см־¹): 1760, 1770 (С=О), 1605 (С=С). Спектр ЯМР ¹Н (DMFA-d7, δ, м.д.): 8.13 уш.с (2Н, 2NH), 7.30 м (5Н, С6Н5), 4.30 д.д.д. (2Н, С9 и С10 , J¹ = J² = 5.4 Гц, J³ = 15.6 Гц), 3.62 с (3Н, СООСН3), 3.48 м (2Н, СН2 бензила), 3.12 м (2Н, 2СН), 2.98 м (2Н, 2СН), 2.75 м (4Н, 2СН, СН2), 2.55 м (2Н, СН2), 2.30 т (2Н, СН2, J 7.34 Гц), 1.75 м (2Н, СН2), 1.65 д.д. (2Н, С11Н2, J¹ = J² = 12.5 Гц). Спектр ЯМР ¹³С (DMFA-d7, δ, м.д.): 174.05 с (С=О), 171.72 с (С=О), 171.60 с (С=О), 140.54 с (С¹ бензила), 132.63 с (-ССl=), 129.19 д (СН бензила), 128.83 д (СН бензила), 127.34 д (СН, бензила), 123.30 с (-CCl=), 51.90 к (ОСН3), 51.59 д (СН), 51.48 д (СН), 49.11 д (СН), 48.70 д (СН), 43.41 т (СН2), 39.24 т (СН2), 29.28 т (СН2), 25.44 т (СН2), 22.91 т (СН2).
9-(4-Метоксикарбонилбутиламид)-3,4,5,6-тетрахлортрицикло-[6.2.1.02,7]ундека-3,5-диен-транс-9α, 10β-дикарбоновой кислоты XIII и его 9β, 10α-изомер XIIIa.
К смеси 1.0 г кислоты XIV в 10 мл ТГФ при перемешивании прибавляли (20ºС) по каплям раствор 0.6 г ДДС в 10 мл ТГФ в течение 30 мин. После образования осадка добавляли 0.45 г гидрохлорида метилового эфира δ-аминовалериановой кислоты и 1.0 г триэтиламина в 10 мл ТГФ. По окончании реакции (ТСХ) ТГФ упарили, массу подкисляли HCl и экстрагировали этилацетатом. Получено 0.6 г (61%) амида с т.пл. 54-58ºС. Анализ ВЭЖХ показал наличие двух изомеров XIII и XIIIa, которые не удалось разделить и использованы в виде смеси в последующих реакциях.
9-Бензиламид-10-(4-Метоксикарбонилбутиламид)-3,4,5,6-тетрахлортри- цикло[6.2.1.02,7]ундека-3,5-диен-транс-9α,10β-дикарбоновой кислоты XII и его 9β,10α-изомер XIIa. По вышеуказанной методике из 400 мг смеси амидов XIII и XIIIa, 150 мг бензиламина и 200 мг ДДС получили 360 мг (63%) смеси бисамидов XII и XIIa в соотношении 1:1, которые разделены ВЭЖХ и охарактеризованы.
Соединение XII: ИК спектр (см־¹): 1760, 1780 (С=О), 1610 (С=С). Спектр ЯМР ¹³С (DMFA-d7, δ, м.д.): 173.62 с (С=О), 172.12 с (С=О), 174.02 с (С=О), 140.40 с (С¹ бензила), 132.89 с (С4), 132.35 с (С5), 128.94 д (СН, бензила), 128.07 д (СН, бензила), 127.44 д (СН, бензила), 123.56 с (С³), 123.31 с (С6), 52.07 к (ОСН3), 51.49 т, 49.88 т, 48.27 т, 48.75 т, 47.05 д, 43.40 д, 39.39 т, 33.74 т, 22.85 д.
Соединение XIIа: ИК спектр (см־¹): 1760, 1780 (С=О), 1610 (С=С). Спектр ЯМР ¹³С (DMFA-d7, δ, м.д.): 174.12 с (С=О), 173.58 с (С=О), 172.48 с (С=О), 140.22 с (С¹ бензила), 132.55 с (С4), 132.12 с (С5), 128.88 д (СН, бензила), 128.03 д (СН, бензила), 127.32 д (СН, бензила), 123.44 с (С³), 123.30 с (С6 ), 51.89 к (ОСН3), 51.44 т, 49.85 т, 48.25 т, 48.66 т, 47.05 д, 43.38 д, 39.33 т, 33.68 т, 22.77 д.
Список литературы
- Hamberg M., Samuelsson B. Detection and isolation of an endoperoxide interme diate in prostaglandin biosyntesis // Proc. Natl. Acad. Sci. USA. - 1973. V.70. N 3. - P. 899.
- Hamberg M., Svensson J., Wakabayashi T., Samuelsson B. Isolation and structure of two prostaglandin endoperoxides that cause platelet aggregation // Proc. Natl. Acad. Sci. - USA. 1974. V.71. No 2. - P. 345.
- Corey E.J., Narasaka K., Shibasaki M. A direct, stereocontrolied total synthesis of the 9,11-azo analogue of the prostaglandin endoperoxide, PGH2 //J. Amer. Chem. Soc. - 1976. V. 98. No 20. - P. 6417.
- Bundy G.L. The synthesis of prostaglandin endoperoxide analogs// Tetrahedron Lett. - 1975. No 24. - P. 1957.
- Trost B.M., Timko J.M., Stantion J.L. An enantioconvergent approach to prostaglandins. // J. Chem. Soc. Chem. Commun. - 1978. No10. - P. 436.
- Corey E.J., Shbasaki M., Nicolaou K.C. et al. Simple, stereocontrolled total synthesis of a biologically activ analog of the prostaglandin endoperoxides (PGH2, PGG2) // Tetrahedron Lett. - 1976. No 10. - P.737.
- Литвинов Р.И. Современные ингибиторы функции тромбоцитов. // Казанский медицинский журнал – 2004, Т. 85. Вып.2 – С. 125.
- Michael R. H., Anastasia P., Helen B. и др. Функциональный полиморфизм простагландинэндопероксид синтазы позволяет предсказать неблагоприятный исход саркоидоза // Amer. J. of Respiratory and Critical Care Medicine – 2006. V. 174. - P. 915.
- Corey E.J., Niwa H., Bloom M., Ramwell P.W. Synthesis of a new prostaglandin endoperoxide (PGH2) analog and its function as a inhibitor of the biosynthesis of tromboxane A2 (TBXA2). // Tetrahedron Lett. - 1979. No 8. - P. 671.
- Kametani T., Suzuki T., Kamada Sh., Unno K. Synthesis of the prostaglandin H2 analogue dl-9,11- ethano -9,11-dideoxaprostaglandin H2. // J. Chem. Soc. Perkin Trans. - 1981. No12. - P. 3101.
- Lieb F., Niewohner U., Wendisch D. 6-(3-Carbamoylbicyclo[2.2.1]hept-2-yl) hexaansauren, eine newe klasse von TxA2 antagonisten. // Lieb. Ann. Chem. – 1987. P. 607.
- Kobayashi T., Tahara Y., Matsumoto M. et al. Roles of thromboxane A(2) and prostacyclin in the development of atherosclerosis in apoE-deficient mice // J. Clin. Invest. - 2004. V.114. - P.784.
- Nakane M., Reid J.A., Han. Wen Ching et al. 7-Oxabicyclo[2.2.1]heptyl carboxylic acid as thromboxane A2 antagonists^ aza ω-chain analogs. // J. Med. Chem. 1990. V.33. No 9. - P. 2465.
- Pat. Eur. № 373950 / C.A. 1991. V.114. 23676m.
- Pat Eur. № 373951 / C.A. 1991. V.114. 6146w.
- Pat. Eur. № 373915 // C.A. 1991. V.114. 6147x
- Исмаилов С.А. Диеновая конденсация 5.5-диметокситетрахлорциклопента-диена с ангидридами эндо- и элзо-бицикло[2.2.1]-гепт-5-ен дикарбоновой-2,3-кислоты и стереоспецифические превращения аддуктов: Автореф. дисс. канд. хим. наук. - Казань, 1980 - 21 с.
- Шнулин А.Н., Исмаилов С.А., Салахов М.С., Мамедов Х.С. Молекулярная и кристаллическая структура ангидрида экзо-экзо-3,4,5,6-тетрахлортри-цикло[6.2.1.02,7]ундека-3,5-диен-9,10-дикарбоновой кислоты // Ж. структ. хим. - 1981. T.22. Bып.3. - С.100.
- Шнулин А.Н., Исмаилов С.А., Салахов М.С. и др. Молекулярная и кристаллическая структура ангидрида экзо-эндо-3,4,5,6-тетрахлортрицикло [6.2.1.02,7]ундека-3,5-диен-9,10-дикарбоновой кислоты // Кристаллография - 1982. T.27. Bып.2. - С.273.