СТРУКТУРА И ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА ВЗРЫВЧАТЫХ TATB, PETN И TATT

Празян Т.Л.
Студент, Кемеровский государственный университет
СТРУКТУРА И ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА ВЗРЫВЧАТЫХ TATB, PETN И TATT
Аннотация
Одним из перспективнейших направлений повышения безопасности взрывных работ, в частности, в горнодобывающей промышленности, является использование технологий лазерного инициирования, способных обеспечить исключение возможности срабатывания системы инициирования в несанкционированных случаях и ее гарантированное срабатывание в санкционированных. В настоящей работе проводятся исследования веществ, соответствующих практическим требованиям. Такими веществами являются TATB, PETN и TATT. Работа раскрывает свойства геометрических и термодинамических свойств данных молекул.
Ключевые слова: TATB, PETN, TATT.
Prazyan T.L.
Student, Kemerovo State University
STRUCTURE AND THERMODYNAMICS PROPERTIES OF EXPLOSIVE TATB, PETN AND TATT
Abstract
One of the most perspective ways in increasing the safety of explosive works, in particular, in coal-mining industry, is the use of laser initiation technologies, which is able to eliminate the possibility of functioning of the system of initiation in unauthorized cases, and its assured functioning in the sanctioned ones. In this work the substances meeting the practical requirements are being researched. Such substances are TATB, PETN and TATT. The research reveals the geometrical and thermodynamics properties of these molecules.
Keywords: TATB, PETN, TATT.
TATB (C6N6O6H6), PETN (C5N4H8O12) и TATT (C3N10H6) относятся к классу энергетических материалов. Главным преимуществом этих веществ является хорошая термостойкость и низкая чувствительность к механическим воздействиям, а также высокая скорость детонации. Эти структуры используют преимущественно в качестве взрывного компонента.
В работе [1] исследованы свойства кристаллического TATB, в том числе исследованы структурные свойства этого вещества классическим потенциалом межмолекулярного взаимодействия. Работа [2] описывает оптимизацию молекулы TATB методом ONIOM(B3LYP/6-311++G**:UFF) с дальнейшим исследованием данного вещества во взаимодействии с другими. [3] посвящена сравнению исследования молекулярного TATB методами MP2 и B3LYP. В [4] проведены исследования TATB при внешнем воздействии на твердое вещество с первоначальным расчетом геометрических параметров молекулы методами LDA и BPW91 и кристалла методом LDA. В работе [5] рассмотрены взаимодействия кластеров TATB, результаты получены с использованием базисов 6-311G(d,p), 6-311G(2d,2p), AUG-cc-pVDZ и методов MP2 и B3LYP.
Работа [6], посвященная изучением колебательных свойств PETN методом DFT, рассмотрена из первых принципов. В [7] проведены исследования PETN при внешнем воздействии на вещество с первоначальным расчетом геометрических параметров молекулы с использованием теории функционала плотности PBE в сочетании с базисным набором 6-31G**. В [8] представлено разложение PETN по энергетическим уровням исходя из оптимизации геометрии методами/базисами MP2/6-31G(d) и ONIOM(CASSCF(6,6)/6-31G(d):UFF), с результатами которой в дальнейшем произведено сравнение. Исследованиям колебательных свойств изолированных молекул и кристаллов PETN посвящена работа [9] методами B3LYP, BLYP, BOP, BP, HCTH, PBE, PW9,1 RPBE, VWN-BP, PWC, VWN.
Структурные и энергетические свойства TATT методом B3LYP с помощью базиса 6-31+G(d,p) исследованы в [10].
Из последних работ, выполненных по данным структурам, можно сделать вывод о том, что молекулярные свойства изучены не полностью. Целью настоящей работы является подробное исследование структурных, колебательных и энергетических свойств молекул TATB и PETN. Изучение молекулы TATT началось недавно и исследование структуры и свойств является актуальным вопросом.
Для исследования данных соединений использовался пакет GAMESS [11], в которых реализован метод теории функционала электронной плотности в комбинации обменного и корреляционного потенциала B3LYP. Данный метод является гибридным. Базис, используемый в пакете GAMESS: n-31G*[12].
Молекула TATB имеет плоскую структуру с симметрией C2h, в то время как молекула PETN имеет пространственную структуру с симметрией S4. Молекула TATT имеет объемную структуру, но симметрия у нее отсутствует. На рисунке 1 представлена визуализация молекул TATB и PETN.
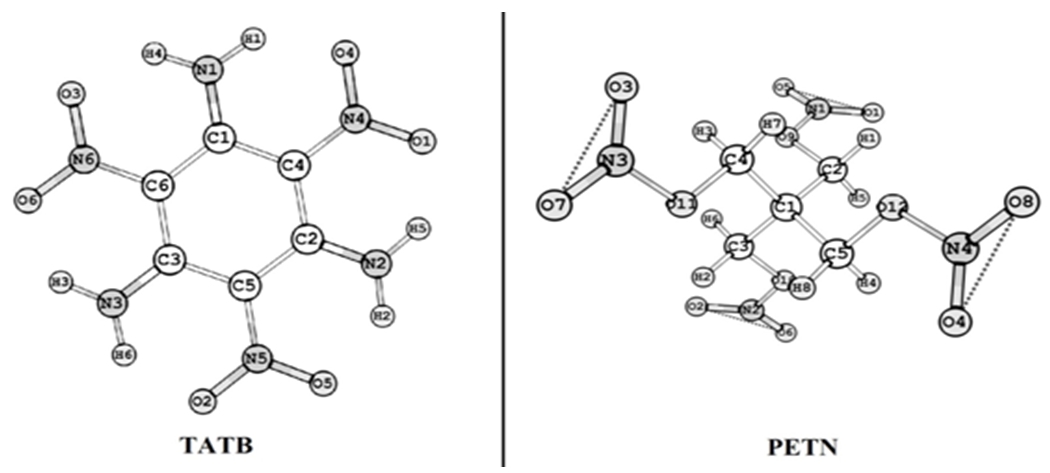
Рис. 1 - Визуализация молекул TATB и PETN
Молекула TATT имеет необычную структуру, отличную от TATB и PETN. В этой молекуле отсутствуют атомы кислорода. На рисунке 2 изображена геометрическая структура молекулы TATT с приведенными длинами связей в единицах Å.
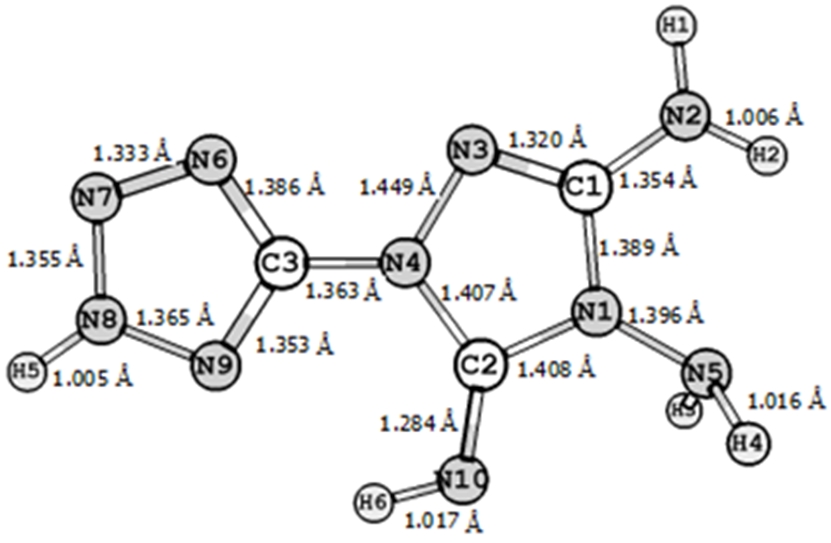
Рис. 2 - Геометрическая структура молекулы TATT
В таблицах 1-4 демонстрируются результаты вычислений, полученные в данной работе в сравнении с последними теоретическими расчетами других авторов, а также экспериментальными данными.
Таблица 1 - Длины связей в молекуле TATB
| Длина связи (Å) | Настоящая работа | [2] | Эксперимент [13] |
| RC1-C4 | 1.44 | 1.45 | 1.44 |
| RC2-N2 (amino) | 1.33 | 1.33 | 1.31 |
| RC4-N4 (nitro) | 1.42 | 1.44 | 1.42 |
| RN4-O1 (RN4-O4) | 1.28 | 1.24 | 1.24 |
| RN2-H5 (RN2-H2) | 1.01 | 1.01 | 1.00 |
Таблица 2 - Углы в молекуле TATB
| Углы (градусы) | Настоящая работа | [2] | Эксперимент [13] |
| α(C–N–H) | 117 | 117 | 122 |
| α(C–N–O) | 121 | 120 | 122 |
| α(H–N–H) | 125 | 125 | 123 |
| α(O–N–O) | 118 | 119 | 118 |
| α(C–C–N) (amino) | 120 | 120 | 120 |
| α(C–C–N) (nitro) | 119 | 119 | 120 |
Как можно видеть из результатов вычислений полученные длины связей и углы находятся в хорошем согласии с экспериментом и не уступают в точности другим теоретическим данным.
Различия между полученными данными оптимизации геометрии молекул TATB и PETN с экспериментальными данными связаны с тем, что экспериментальные данные были получены в кристаллах, в то время как в настоящей работе рассматриваются отдельные молекулы, но преимущество изучения отдельных молекул в удобном представлении и изучении их пространственного строения.
Таблица 3 - Длины связей в молекуле PETN
| Длина связи (Å) | Настоящая работа | [9] | Эксперимент [9] |
| RC1-C2 | 1.54 | 1.54 | 1.53 |
| RC2-H1 (RC2-H5) | 1.08 | 1.09 | 0.99 |
| RC2-O9 | 1.47 | 1.44 | 1.45 |
| RN1-O5 (RN1-O1) | 1.23 | 1.21 | 1.20 |
| RN1-O9 | 1.48 | 1.43 | 1.40 |
Таблица 4 - Углы в молекуле PETN
| Углы (градусы) | Настоящая работа | [9] | Эксперимент [9] |
| C1_C2_O9 | 105.3 | 106.3 | 106.5 |
| C2_O9_N1 | 113.4 | 113.6 | 112.6 |
| O9_N1_O1 | 116.7 | 116.9 | 118.1 |
| O9_N1_O5 | 112.4 | 112.3 | 112.1 |
| O1_N1_O5 | 130.8 | 130.8 | 129.8 |
Энергия, необходимая электрону для его отрыва с внешней остовной орбитали в молекуле TATB, равна 4.36 эВ. Энергия, которую необходимо сообщить электрону для его отрыва с внешней остовной орбитали в молекуле PETN, равна 6.78 эВ. Из полученных значений можно сделать вывод о том, что ширина между внешним и возбужденным состояниями электронов в TATB меньше, чем в PETN. В то же время ширина между внешним и возбужденным состояниями электронов в TATT равна 4.30 эВ.
Потенциал B3LYP более приемлем для подсчета термодинамических параметров исследуемых молекул. Сводная таблица по термодинамическим функциям представлена в таблице 5.
Таблица 5 - Термодинамические функции TATB, PETN, TATT
| E | H | G | S | |
| Молекула | кДж/Моль | кДж/Моль | кДж/Моль | Дж/Моль-К |
| TATB | 449.877 | 452.356 | 331.546 | 405.197 |
| PETN | 540.942 | 543.421 | 408.442 | 452.721 |
| TATT | 391.528 | 394.007 | 275.798 | 396.474 |
Как видно из таблицы, внутренняя энергия в молекуле TATT меньше внутренних энергий в молекулах TATB и PETN. То же можно сказать об остальных термодинамических функциях.
Данная работа, направленная на подробное рассмотрение структурных, энергетических и колебательных свойств TATB, PETN и TATT, в полной мере раскрывает характеристики выбранных к изучению взрывчатых веществ. Геометрические характеристики молекул TATB, PETN и TATT получены из первых принципов гибридным методом B3LYP, а приближенное соответствие характеристик молекул TATB и PETN к экспериментальным данным подтверждает правильность выбранного метода и базиса для расчета иных свойств исследуемых молекул. Приведенные в таблице 5 функции (внутренняя энергия, энтальпия, потенциал Гиббса и энтропия) изучаемых термодинамических систем достаточно полно описывают количественные термодинамические свойства этих систем.
Список литературы
Gee, R.H. Ab initio based force field and molecular dynamics simulations of crystalline TATB / R.H. Gee, Roszak, K. Balasubramanian, L.E. Fried // J. Phys. Chem. – 2004. – V 120. – № 15. – P. 1-8.
Wang, L. Adsorption of the insensitive explosive TATB on singlewalled carbon nanotubes / Wang, C. Yi, H. Zou, H. Gan, J. Xu, W. Xu // Molecular Physics. – 2011. – V 109. – № 14. – P. 1841-1849.
Manaa, M. R. Internal Rotation of Amino and Nitro Groups in TATB: MP2 Versus DFT (B3LYP) / M. R. Manaa, L.E. Fried, R.H. Gee // Phys. Chem. – 2002. – № 106. – P. 8806-8810.
Wu, С. J. Electronic structure of solid 1,3,5-triamino-2,4,6-trinitrobenzene under uniaxial compression: Possible role of pressure-induced metallization in energetic materials / С. J. Wu, L.H. Yang, E. Fried, J. Quenneville, T.J. Martinez // Physical review. – 2003. – № 235101. – P. 1-7.
Roszak, S. Molecular interactions of TATB clusters / S. Roszak,H. Gee, K. Balasubramanian, L.E. Fried // Chemical Physics Letters. – 2003. – № 374. – P. 286–296.
Perger, W. F. First-principles study of pentaerythritol tetranitrate single crystals under high pressure: vibrational properties / W. F. Perger, J. Zhao, J. M. Winey, Y. M. Gupta // Chemical Physics Letters. – 2006. – № 428. – 394-399.
Gan, C. K. All-electron density-functional studies of hydrostatic compression of pentaerythritol tetranitrate C(CH2ONO2)4 - Los Alamos: Los Alamos National Laboratory / K. Gan, T. D. Sewell, M. Challacombe // The American Physical Society. – 2004. – V 69. – P. 1-7.
Yu, Z. Decomposition of pentaerythritol tetranitrate [C(CH2ONO2)4] following electronic excitation / Z. Yu, E. R. Bernstein // Phys. Chem. – 2011. – V 135. - № 154305. – P. 1-11.
Allis, D. G. Theoretical Analysis of the Terahertz Spectrum of the High Explosive PETN / D. G. Allis, T. M. Korter // Phys. Chem. – 2006. – № 7. – P. 2398 – 2408.
Tao, G.-H. A thermally stable nitrogen-rich energetic material—3,4,5-triamino-1-tetrazolyl-1,2,4-triazole (TATT) / G.-H. Tao, B. Twamley, J. M. Shreeve // J. Mater. Chem. – 2009. – V19. – P. 5850-5854.
Schmidt, M.W. General Atomic and Molecular Electronic Structure System / M.W. Schmidt, K.K. Baldridge, J.A. Boatz, S.T. Elbert, M.S. Gordon, J.H. Jensen, S. Koseki, N. Matsunaga, K.A. Nguyen, S. Su, T.L. Windus, M. Dupuis, J.A. Montgomery // J. Comput. Chem. – 1993. - V 14. - P. 1347-1363.
Basis Set Exchange [Электронный ресурс] URL: https://bse.pnl.gov/bse/portal (дата обращения 8.10.2013).
Cady, H. H. The crystal structure of 1,3,5-triamino-2,4,6-trinitrobenzene / H. H. Cady, A. C. Larson // Acta Cryst. – 1965. – V 18. – P. 485-496.
Список литературы
Gee, R.H. Ab initio based force field and molecular dynamics simulations of crystalline TATB / R.H. Gee, Roszak, K. Balasubramanian, L.E. Fried // J. Phys. Chem. – 2004. – V 120. – № 15. – P. 1-8.
Wang, L. Adsorption of the insensitive explosive TATB on singlewalled carbon nanotubes / Wang, C. Yi, H. Zou, H. Gan, J. Xu, W. Xu // Molecular Physics. – 2011. – V 109. – № 14. – P. 1841-1849.
Manaa, M. R. Internal Rotation of Amino and Nitro Groups in TATB: MP2 Versus DFT (B3LYP) / M. R. Manaa, L.E. Fried, R.H. Gee // Phys. Chem. – 2002. – № 106. – P. 8806-8810.
Wu, С. J. Electronic structure of solid 1,3,5-triamino-2,4,6-trinitrobenzene under uniaxial compression: Possible role of pressure-induced metallization in energetic materials / С. J. Wu, L.H. Yang, E. Fried, J. Quenneville, T.J. Martinez // Physical review. – 2003. – № 235101. – P. 1-7.
Roszak, S. Molecular interactions of TATB clusters / S. Roszak,H. Gee, K. Balasubramanian, L.E. Fried // Chemical Physics Letters. – 2003. – № 374. – P. 286–296.
Perger, W. F. First-principles study of pentaerythritol tetranitrate single crystals under high pressure: vibrational properties / W. F. Perger, J. Zhao, J. M. Winey, Y. M. Gupta // Chemical Physics Letters. – 2006. – № 428. – 394-399.
Gan, C. K. All-electron density-functional studies of hydrostatic compression of pentaerythritol tetranitrate C(CH2ONO2)4 - Los Alamos: Los Alamos National Laboratory / K. Gan, T. D. Sewell, M. Challacombe // The American Physical Society. – 2004. – V 69. – P. 1-7.
Yu, Z. Decomposition of pentaerythritol tetranitrate [C(CH2ONO2)4] following electronic excitation / Z. Yu, E. R. Bernstein // Phys. Chem. – 2011. – V 135. - № 154305. – P. 1-11.
Allis, D. G. Theoretical Analysis of the Terahertz Spectrum of the High Explosive PETN / D. G. Allis, T. M. Korter // Phys. Chem. – 2006. – № 7. – P. 2398 – 2408.
Tao, G.-H. A thermally stable nitrogen-rich energetic material—3,4,5-triamino-1-tetrazolyl-1,2,4-triazole (TATT) / G.-H. Tao, B. Twamley, J. M. Shreeve // J. Mater. Chem. – 2009. – V19. – P. 5850-5854.
Schmidt, M.W. General Atomic and Molecular Electronic Structure System / M.W. Schmidt, K.K. Baldridge, J.A. Boatz, S.T. Elbert, M.S. Gordon, J.H. Jensen, S. Koseki, N. Matsunaga, K.A. Nguyen, S. Su, T.L. Windus, M. Dupuis, J.A. Montgomery // J. Comput. Chem. – 1993. - V 14. - P. 1347-1363.
Basis Set Exchange [Электронный ресурс] URL: https://bse.pnl.gov/bse/portal (дата обращения 8.10.2013).
Cady, H. H. The crystal structure of 1,3,5-triamino-2,4,6-trinitrobenzene / H. H. Cady, A. C. Larson // Acta Cryst. – 1965. – V 18. – P. 485-496.