ИЗУЧЕНИЕ ЭКСПРЕССИИ РЕЦЕПТОРА РОСТОВОГО ФАКТОРА И СТРУКТУРЫ ГЕНА ТРАНСФОРМИРУЮЩЕГО ФАКТОРА РОСТА ПРИ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ

Пушкарева А.Э.1, Хусаинова Р.И.2, Валиев Р.Р.3, Хуснутдинова Э.К.4
1 доцент кафедры госпитальной терапии, кандидат медицинских наук, ФГБОУ ВПО «Башкирский Государственный Медицинский Университет» Министерства здравоохранения и социального развития РФ, г. Уфа; 2 старший научный сотрудник лаборатории молекулярной генетики человека, доктор биологических наук, ФГБУН «Институт биохимии и генетики» Уфимского научного центра РАН, г. Уфа; 3 заведующий лабораторией ПЦР-анализа, кандидат биологических наук, ФГБОУ ВПО «Башкирский государственный университет», г. Уфа; 4 доктор биологических наук, профессор, заведующий отделом геномики, ФГБУН «Институт биохимии и генетики» Уфимского научного центра РАН, г. Уфа, заведующий кафедрой генетики и фундаментальной медицины, ФГБОУ ВПО «Башкирский государственный университет», г. Уфа
ИЗУЧЕНИЕ ЭКСПРЕССИИ РЕЦЕПТОРА РОСТОВОГО ФАКТОРА И СТРУКТУРЫ ГЕНА ТРАНСФОРМИРУЮЩЕГО ФАКТОРА РОСТА ПРИ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ
Аннотация
Проведены иммуногистохимическое исследование аутопсийной ткани миокарда у больных ХСН с выраженной гипертрофией левого желудочка (ЛЖ) и дилатацией полостей сердца с использованием моноклональных мышиных анти-человеческих антител, афинносвязывающихся с рецепторами к трансформирующему фактору роста бета (TGFβR1) и анализ 9 экзонов гена рецептора трансформирующего фактора роста бета (TGFВR1) у больных с хронической сердечной недостаточностью (ХСН) с различными типами ремоделирования. Обнаружено достоверное увеличение экспрессии TGFβ-R1 у больных гипертонической болезнью с выраженной ГЛЖ и обнаружена положительная корреляция между экспрессией TGFβR1 и гипертрофией кардиомиоцитов. Выявлены две миссенс-мутации, одна из которых обнаружена впервые, две синонимичные замены и полиморфный вариант сайта сплайсинга.
Ключевые слова: хроническая сердечная недостаточность, экспрессия рецептора трансформирующего фактора роста бета, ремоделирование сердца, гипертрофия левого желудочка, мутации в гене рецептора трансформирующего фактора роста бета
Pushkareva A.E.1, Khusainova R.I.2, Valiev R.R.3, Khusnutdinova E.K.4
1 MD, associate professor, The Bashkir State Medical University, Ufa; 2 Senior Researcher, Doctor of Biology, Laboratory of Molecular Human Genetics, Institute of Biochemistry and Genetics, Ufa Science Centre of the Russian Academy of Sciences, Ufa; 3 Head of PCR-analysis Laboratory, PhD in Biology, The Bashkir State University; 4 Head of the Department of Genomics, Doctor of Biology, professor, Institute of Biochemistry and Genetics, Ufa Science Centre of the Russian Academy of Sciences, The Bashkir State University, Ufa
THE STUDY OF GROWTH FACTOR RECEPTOR EXPRESSION AND GENE STRUCTURE OF THE TRANSFORMING GROWTH FACTOR IN HEAD FAILURE
Abstract
The Immunohistochemical study of autopsy myocardial tissue performed for heart failure patients with severe left ventricular hypertrophy (LVH) and dilatation of the heart using monoclonal mouse anti-human antibodies, affinity binding to receptors of transforming growth factor (TGFβR1) and analyzed 9 exons of the transforming growth factor receptor gene (TGFBR1) in heart failure patients with various types remodeling. We found a significant increase of TGFβR1 expression in hypertension patients with severe LVH and positive correlation between TGFβR1 expression and hypertrophy of cardiomyocytes. Identified two missense mutations, one of them for the first time, two synonymous substitutions and polymorphic variant of splicing site.
Keywords: chronic heart failure, expression of receptor of the transforming body height factor beta, remodeling of heart, hypertrophy of left ventricle, mutation in a gene of a receptor of the transforming body height factor beta
Введение
Трансформирующий фактор роста бета (TGF-β) – представитель семейства многофункциональных цитокинов, регулирующих разнообразные клеточные функции, такие как рост, адгезию, миграцию, апоптоз, пролиферацию и дифференциацию [1]. Нарушения TGF-β сигнального пути обнаружены при многих заболеваниях человека: онкологических, сердечно-сосудистых, наследственных заболеваниях соединительной ткани. Мутации, затрагивающие TGF-сигнальные пути были выявлены у пациентов с артериальными аневризмами, пороками сердца, кардиомиопатиями, наследственными геморрагическими телеангиэктазиями и т.д. [2, 3, 4, 5]. В экспериментах invitro с использованием культивированных тканей сердца показана важность этого сигнального пути для создания мезенхимальных клеток, которые, в конечном счете, способствуют развитию клапанов и перегородок зрелого сердца [6]. У млекопитающих были выявлены три изоформы TGF-β (TGF-β1, TGF-β2 и TGF-β3), TGFβ1 является основным фактором ремоделирования сердца за счет активации разрастания волокнистой соединительной ткани (фиброза) и гипертрофии кардиомиоцитов. Показано, что уровень TGFβ1 коррелирует со степенью фиброза [7,8]. Несколько исследований выявили вовлеченность TGFβ1 в патологическое развитие фиброза [8,9,10]. В патогенезе хронической сердечной недостаточности (ХСН) важное место занимает процесс фиброза, приводящее к нарушению функции миокарда и ремоделирования сердца. У пациентов с гипертрофической и дилатационной кардиомиопатиями обнаружен повышенный уровень TGFβ1 в миокарде [10, 11, 12].
Учитывая неперекрывающийся [13] и многофункциональный характер TGFβ сигнальных путей (десятки фенотипов среди TGFβ нокаутированных мышей) [14], существуют многочисленные потенциальные механизмы, которые могут участвовать в развитии сердечно-сосудистых заболеваний [15].
Рецептор TGFB первого типа (TGFВR1) является ключевым звеном TGFβ сигнального пути, контролирующего морфогенез тканей. Ген TGFВR1 локализован на хромосоме 9q22, составляет приблизительно 31 тысяч пар оснований в длину и состоит из девяти экзонов [16].
За последние 25 лет произошел значительный прогресс в понимании молекулярного патогенеза, генетических причин сердечно-сосудистых заболеваний, а также в разработке более эффективных способов лечения. Эти достижения привели к снижению смертности от болезней сердца, особенно, в промышленно развитых странах. Несмотря на это, распространенность и заболеваемость сердечной недостаточностью остаются поразительно высокими, что подтверждает актуальность поиска новых молекулярно-генетических маркеров диагностики, профилактики и лечения заболеваний сердечно-сосудистой системы.
Целью исследования является изучение экспрессии рецептора TGF β первого типа (TGFβR1) и анализ нуклеотидной последовательности гена TGFBR1 у больных с хронической сердечной недостаточностью (ХСН), поиск связи идентифицированных изменений с различными типами ремоделирования сердца.
Материал и методы исследования
Материалом для исследования послужили образцы ДНК 207 больных ХСН на фоне ИБС, дилатационной кардиомиопатии, артериальной гипертонии (II –III стадии согласно Российским рекомендациям, 2004), гипертрофической кардиомиопатии без признаков обструкции выносящего тракта ЛЖ и их сочетания; четкие клинические признаки II – IV ФК ХСН (NYHA, 1994) моложе 60 лет. Критериями исключения из исследования были: стеноз клапанных отверстий, врожденные пороки сердца, в период острой левожелудочковой недостаточности, острый коронарный синдром, сахарный диабет, дефекты опорно-двигательного аппарата. В зависимости от типа ремоделирования ЛЖ группа больных была разделена на 2 группы: 1 группа – 84 больных с признаками эксцентрической гипертрофии ЛЖ (ОТС менее 0.45, увеличение ИММЛЖ, дилатация ЛЖ, фракция выброса 20-45% по Simpson); 2 группа – 123 больных с признаками концентрической гипертрофии ЛЖ (ОТС более 0.45 и увеличение ИММЛЖ, выраженная гипертрофия стенок ЛЖ более 1.5 см). Эхокардиографические измерения проводились в двухмерном режиме на аппарате Vingmed 5 (Норвегия). Толщина стенок ЛЖ и размеры полости определялись из парастернальной позиции длинной оси левого желудочка. Масса миокарда ЛЖ (ММЛЖ) вычислялась на основании показателей его длины и толщины по короткой оси из парастернального доступа по формуле R.Devereux и N.Reicheck. Индекс массы миокарда ЛЖ (ИММЛЖ) рассчитывали как отношение массы миокарда к площади поверхности тела. За нормальные значения ИММЛЖ принимались цифры менее 134 г/м2 для мужчин и 110 г/м2 для женщин [2]. ММЛЖ считалась нормальной при значениях менее 215г. Относительная толщина стенок ЛЖ определялась следующим способом: ОТС=(ТМЖП+ТЗСЛЖ)/КДРЛЖ, где ТМЖП – толщина межжелудочковой перегородки ЛЖ, ТЗСЛЖ – толщина задней стенки ЛЖ, КДРЛЖ – конечный диастолический размер ЛЖ. За повышение ОТС принимались значения 0,45 и более.
В качестве контрольной группы использованы 188 индивидов без признаков заболеваний сердечно-сосудистой системы (ССС). Все участники исследования подписали информированное согласие, работа получила одобрение биоэтического комитета ИБГ УНЦ РАН и БГМУ.
Геномную ДНК выделяли методом фенольно-хлороформной экстракции из лейкоцитов венозной крови [17]. Амплификацию экзонов гена TGFBR1 проводили с помощью праймеров, описанных ранее, а также сконструированных самостоятельно в программе PrimerSelect (DNASTAR, США) (табл.1).
Поиск изменений нуклеотидной последовательности в гене TGFBR1проводили методом анализа конформационного полиморфизма однонитевой ДНК (SSCP) по методике, предложенной Орита М. с соавт. с щелочной и температурной денатурацией [18]. После денатурации образцы наносили на 8% полиакриламидный гель (ПААГ). Электрофорез геля длиной 20 см, толщиной 1 мм проходил при комнатной температуре при напряжении 100В в течение 20 - 40 часов. Окраска геля проводилась в течение 25 минут 0,09% раствором азотнокислого серебра (AgNO3). Анализ образцов проводился по критерию присутствия или отсутствия дополнительных полос по сравнению с контрольной ДНК (норма). Определение последовательности нуклеотидов у образцов с измененной подвижностью однонитевой ДНК проводили с помощью автоматического секвенатора ABIPRISM модель 310 («AppliedBiosystems») с использованием набора для флюорисцентного мечения DYEnamicTMET, согласно протоколу фирмы производителя («AmershamPharmaciaBiotech» DYEnamicTMETTerminatorCycleSequencingKit). Для «прочтения» последовательности нуклеотидов использовали приложение BioEditv.5.0.9. (1997- 2001), а для анализа полученных сиквенсов - MegAlign из пакета программ DNAStarInc (1993-2002).
Также были исследованы 101 образцов аутопсийного материала, 81 мужчина и 20 женщин в возрасте от 32 до 60 лет, средний возраст (48,3±1,8). Больные ИБС с развитием сердечной недостаточности ФК III-IV составили 1-ую группу 33 человека, во 2-ую группу из 27 человек вошли больные гипертонической болезнью и выраженной гипертрофией ЛЖ (более 20мм), 3-ю группу составили 25 больных дилатационной кардиомиопатией и сердечной недостаточностью ФК III-IV, 4-ая – контрольная группа (16 человек) без признаков поражения сердца, сопоставимые по возрасту и полу с группами сравнения. Из каждого секционного препарата (миокарда левого желудочка) вырезали 4-8 кусочков длиной и шириной до 1 см, толщиной до 0,5 см. Биоптаты не брали в зонах крупноочагового кардиосклероза. Кусочки фиксировали в 10% нейтральном формалине 14-16 часов. После проводки спирт-ксилол заливали в парафин торговой марки Histomixplus. Изготавливали парафиновые срезы толщиной 3-5 мкм. В качестве обзорных гистологических окрасок использовали гематоксилин и эозин, пикрофуксин.
Для иммуногистохимического исследования были использованы моноклональные мышиные анти-человеческие антитела, производимые корпорацией NovoCastra, афинносвязывающиеся с рецепторами к трансформирующему фактору роста (TGFβ-R1).
В качестве системы детекции применялась полимеризованная система EnVision+ System-HRP (DAB), для использования с мышиными первичными антителами.
Для полуколичественного анализа результатов реакций был использован визуально-синкретический метод оценки экспрессии по интенсивности проявления DAB-метки: светло-коричневый – 1 балл, коричневый – 2 балла, темно-коричневый – 3 балла. Обработку результатов исследования проводили с помощью пакета программы статистического анализа "SPSSv.13" [SPSSInc., Chicago, Illinois].
РЕЗУЛЬТАТЫ
При изучении экспрессии TGFβ-R1 нами выявлено достоверное увеличение экспрессии TGFβ-R1 у больных гипертонической болезнью с выраженной ГЛЖ в сравнении с группой контроля (р<0,00275), кроме того отмечена отчетливая тенденция по преобладанию экспрессии TGFβ-R1 при гипертонической болезни в сравнении с ИБС, хотя различия не достигают достоверных значений (р<0.064) (рис. 6). Корреляционный анализ выявил достоверную положительную корреляцию между экспрессией TGFβR1 и гипертрофией кардиомиоцитов у больных гипертонической болезнью и выраженной ГЛЖ, такая же корреляционная зависимость определялась у больных дилатационной кардиомиопатией, а в группе больных ИБС экспрессия TGFβR1 достоверно коррелировала с толщиной миокарда ЛЖ, следует отметить, что для лиц с неизмененным сердцем таких корреляционных зависимостей для TGFβR1 мы не обнаружили. Учитывая явную вовлеченность рецептора данного ростового фактора в процесс гипертрофии миокарда ЛЖ и фиброзирования миокарда, нам показалось необходимым изучить структурные особенности гена TGFBR1 у больных ХСН.
Проведен анализ нуклеотидной последовательности девяти экзонов и фланкирующих интронных областей гена рецептора трансформирующего фактора роста β первого типа (TGFBR1) в группах больных с ХСН с разными типами ремоделирования ЛЖ. Изменения подвижности однонитевой ДНК обнаружены в 3, 6 и 8 из 9 экзонов исследуемого гена. В экзонах 1, 2, 4, 5, 9 гена TGFBR1не выявлено изменений подвижности при SSCP анализе. Последующее секвенирование образцов с измененной подвижностью одноцепочечной ДНК позволило идентифицировать 5 типов изменения нуклеотидной последовательности (табл. 2, 3).
В 3-ом экзоне обнаружены два типа изменения подвижности одноцепочечной ДНК у двух больных ХСН. Идентифицирована замена гуанина на аденин в 457 положении кодирующей области гена (с.457G>А) в гетерозиготном состоянии у больного с эксцентрическим типом ремоделирования ЛЖ. Мутация подтверждена ПДРФ анализом с помощью рестриктазы HpHI, подбор которого проведен с использованием пакета компьютерных программ Lasergene 5.05 (1989-2002, “DNASTARInc.”, USA) (рис.1).
В результате данной транзиции происходит замена аминокислоты валин на изолейцин в 153 положении белка TGFβR1. Мутация V153I локализована в цитоплазматическом домене белка рецептора TGFβ первого типа, близко к трансмембранному домену и, предположительно, может влиять на конформационное расположение рецептора в клеточной мембране. При этом обе аминокислоты среднего размера, имеют схожие физико-химические свойства, являются гидрофобными, что предполагает нейтральный характер изменения. По данным проекта «1000 геномов» изменение с.457G>А (р.V153I, rs56014374) выявлена у 2 из 1090 изученных индивидов в гетерозиготном состоянии, что составило <0,001% (http://www.1000genomes.org). Оба носителя были европейского происхождения, в популяциях Африки, Азии и Америки изменение с.457G>А в гене TGFβR1 не обнаружен. Замена V153I также была обнаружена в опухолевых клетках одного больного раком молочной железы, функциональная роль данного изменения в канцерогенезе не была определена [19].
В третьем экзоне гена TGFBR1 выявлена ранее неописанная замена аденина на гуанин в 516 положении ДНК, не приводящая к изменению аминокислотной последовательности рецептора TGFβ первого типа (S172S) у больной с выраженной концентрической гипертрофией стенок левого желудочка (рис. 4). В доступной литературе и в базе данных по мутациям случаев выявления мутации с.464А>G (р.H155R) в гене TGFBR1 не обнаружено (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=TGFβR1).
Обнаружена синонимичная замена аденина на цитозин в 1125 положении гена, не приводящая к замене кодируемой аминокислоты (p.Y377Y). В нашем исследовании обнаружен только один случай гетерозиготного носительства с.1125А>С у больного с концентрической формой ГЛЖ умершего в 46 лет от остановки сердечной деятельности. Сердце увеличено в размерах, толщина стенок левого желудочка 2,0 см, дистрофия кардиомиоцитов. При иммуногистохимическом исследовании выявлена повышенная экспрессия TGFβR1, равная 3 баллам.
Ранее данное изменение было выявлено у больных с заболеваниями соединительной ткани из Европы и идентифицировано как полиморфный вариант гена TGFBR1, не влияющий на его функцию [20].В рамках проекта «1000 геномов» полиморфный вариант с.1125А>С (rs7861780) гена TGFBR1 выявлен у европейцев и в смешанной популяции американцев с частотой менее 1% и не обнаружен в популяциях Африки и Азии. При этом выявлены случаи как гетерозиготного носительства (АС), так и гомозиготные индивиды (АА и СС) (http://www.1000genomes.org).
В нашем исследовании в восьмом экзоне гена TGFBR1 выявлена ранее неописанная миссенс-мутация с.1285А>С, приводящая к замене тирозина на серин в 229 позиции белка (Y229S) (рис. 2). Мутация расположена в киназном домене белкаTGFβR1. Изменение затрагивает эволюционно консервативный участок и может привести к нарушениям проводимости сигнала и функции рецептора TGFβ1. Больной умер от острого инфаркта в возрасте 61 года, сердце увеличено в размерах, толщина стенок левого желудочка 2,0 см., миокард вне зоны инфаркта с прослойками соединительной ткани, выявлена дистрофия кардиомиоцитов. Экспрессия TGFβ-R1 составила 2,5 балла. Необходимы дальнейшие исследования для подтверждения функциональной роли мутации Y229S в развитии сердечно-сосудистых заболеваний.
Нами также обнаружена ранее описанная в литературе трансверсия G на А в +24 позиции донорного сайта сплайсинга, расположенного в 7 интроне генаTGFB-R1 (c.1024+24G>A, rs334354) (рис. 5). Функциональная роль данного полиморфизма до конца не выяснена, имеются публикации об ассоциации гомозиготного генотипа *А*А с повышенным риском развития рака молочной железы и прямой кишки [21]. Учитывая высокую частоту данного полиморфизма у больных ХСН и для выяснения его функциональной значимости мы провели анализ полиморфного варианта c.1024+24G>A генаTGFB-R1в группе здоровых индивидов. Выявлено схожее распределение частот аллелей и генотипов исследуемого локуса в общей выборке больных и контроля (табл. 3).
Наблюдается повышение частоты аллеля *А у больных ХСН (0,234) по сравнению с контрольной группой, при этом у мужчин сохраняется схожая тенденция, тогда как у женщин с ХСН наблюдается снижение частоты аллеля *А (0,216) по сравнению с контрольной группой (0,323), различия не достигают статистической значимости (χ2=2,79; р=0,095).
При разделении выборки больных в зависимости от типа ремоделирования ЛЖ частота аллеля *А варьировала от 0,143 у женщин с эксцентрической формой ремоделирования ЛЖ до 0,247 у мужчин с концентрической ГЛЖ. У больных с эксцентрической ГЛЖ наблюдается увеличение частоты предкового аллеля *G по сравнению с группой больных с концентрической ГЛЖ, как в целом, так и при рассмотрении выборок больных в зависимости от пола (табл.3).
Гетерозиготный генотип *A*G чаще всего встречался у женщин из контрольной группы (0,595), самая низкая частота выявлена у женщин с эксцентрической ГЛЖ (0,286), различия не достигают статистической значимости из-за малочисленности выборки больных с эксцентрической ГЛЖ (χ2=2,5; р=0,113). Гомозиготный генотип *А*А не обнаружен у женщин с ХСН, наибольшая частота выявлена у мужчин с концентрической ГЛЖ (0,043). Статистически значимых различий в распределении частот генотипов между группами сравнения не обнаружено. Таким образом, полиморфизм c.1024+24 G>A (rs334354) в 7 интроне генаTGFβ-R1 вероятно, не играет значимой роли в патогенезе ХСН с разными типами ремоделирования ЛЖ.
ОБСУЖДЕНИЕ
В исследованиях китайских ученых по сравнительному изучению экспрессии матриксной металлопротеиназы 9 (ММП 9) и трансформирующего фактора роста β1 (TGFβ1) и его рецептора у больных ИБС было определено, что при наличии нестабильной бляшки экспрессия ММП 9 была значительно выше, а при наличии стабильной бляшки, напротив, достоверно выше оказалась экспрессия TGFβ1 и его рецептора (TGFβR1). Полученные данные позволяют предположить, что TGFβ1 стимулирует продукцию экстрацеллюлярного матрикса, подавляет активность протеиназ и матриксных металлопротеиназ, включая ММП9 и усиливает синтез коллагена, выполняя роль фактора «стабилизации бляшки» [22]. Также было установлено существенное различие в уровне экспрессии TGFβ1 и его рецептора TGFβR1 у пациентов с идиопатической и вторичной формами легочной артериальной гипертензии. Полученные различия в группах по соотношению экспрессия TGFβ1/экспрессия TGFβR1 свидетельствуют о заинтересованности этого цитокина в патогенезе легочной артериальной гипертензии, протекающей с гипертрофией фибробластов, гладкомышечных и эндотелиальных клеток, а также с возрастанием объема экстрацеллюлярного матрикса легочных прекапиллярных артериол [23]
Экспрессия TGFβR1 активно происходит на этапе «гипертрофического ответа» миокарда при ремоделировании сердца у больных эссенциальной гипертонией. Схожая тенденция отмечена и при дилатационной кардиомиопатии.
Есть существенные доказательства предполагать, что трансформирующий фактор роста β является активным триггером фиброзных изменений в гипертрофированном сердце. Уровень экспрессии TGFβ1 в миокарде больных обструктивной гипертрофической кардиомиопатией оказался в 2,5 раза выше, чем у пациентов с негипертрофированным сердцем [24]. Ранее теми же исследователями было выявлено, что аффинность TGFβ1 к своему рецептору была максимальной у индивидуумов с идиопатической гипертрофической кардиомиопатией [25]. Подобные исследования способствуют не только пониманию роли ростовых факторов в ремоделировании миокарда, но и разработкеновых «таркетных» препаратов для коррекции данных процессов. У мышей с постинфарктным ремоделированием в эксперименте применяли специфический ингибитор TGFβR1 (SD 208). Этот гипотетический препарат SD 208 при применении после экспериментального инфаркта миокарда у мышей через 30 дней подтвердил прямое участие TGFβ1 в ангиотензин II-индуцированной гипертрофии миокарда [26].
Таким образом, впервые проведен анализ гена TGFBR1 и уровня экспрессии рецептораTGFβR1 у больных с ХСН из Республики Башкортостан. Выявлено достоверное увеличение экспрессии TGFβ-R1 у больных гипертонической болезнью с выраженной ГЛЖ и обнаружена положительная корреляция между экспрессией TGFβR1 и гипертрофией кардиомиоцитов.
Выявлены две миссенс-мутации в гене TGFBR1, одна из которых обнаружена впервые, две синонимичные замены, мутация сдвига рамки считывания и полиморфный вариант сайта сплайсинга. Полученные результаты указываютна непосредственное участие TGFβR1 и его генетической структуры в процессах ремоделирования миокарда при сердечной недостаточности, а также обосновывают необходимость дальнейших исследований в этом направлении.
Таблица 1 - Последовательности праймеров и характеристика продуктов амплификации

Таблица 2 - Характеристика пациентов ХСН с идентифицированными изменениями в гене TGFBR1
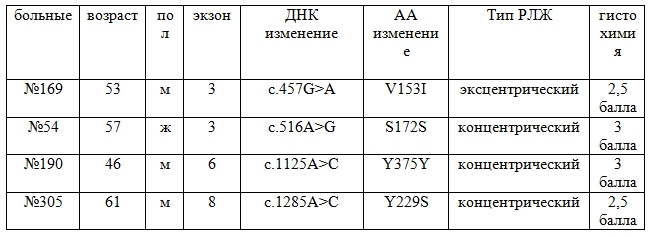
Таблица 3 - Распределение частот аллелей и генотипов полиморфизма c.1255+24G>A(rs334354) генаTGFBR1у больных ХСН и в контрольной группе
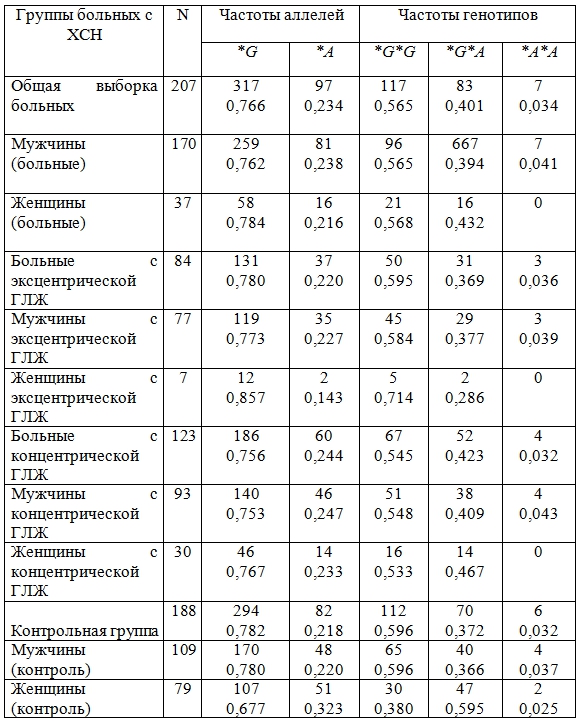
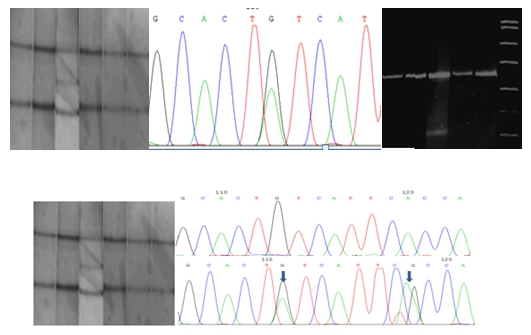
Рис. 1. Идентификация мутаций c.457G>A (V153I) и c.464А>G (H155R) в генеTGFBR1в гетерозиготном состоянии.

Рис. 2. Идентификация миссенс-мутации c.1285A>C (Y229S) в генеTGFBR1в норме (верхний правый) и гетерозиготном состояниях (нижние)
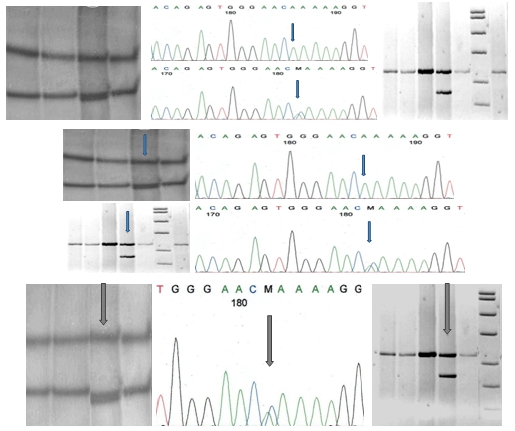
Рис. 3. Выявление методом SSCP анализа, идентификация полиморфного варианта c.1125A>C (Y375Y) генаTGFBR1и подтверждение с использованием фермента рестрикции PspN4I.
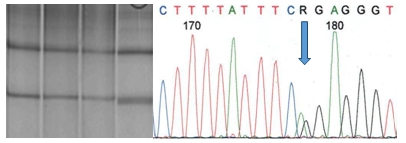
Рис. 4. Идентификация полиморфного варианта c.516A>G (S172S) генаTGFBR1в гетерозиготном состоянии.
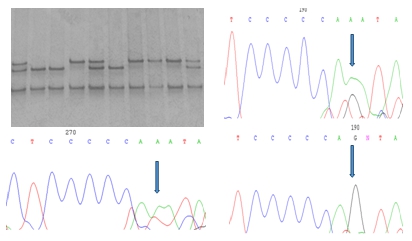
Рис. 5. Идентификация полиморфного варианта c.1024+24G>A(rs334354) генаTGFBR1в гетерозиготном (верхний правый) и гомозиготном состояниях (нижние).
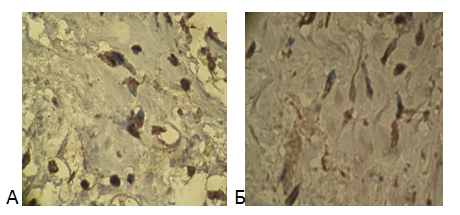
Рисунок 6. Экспрессия рецепторов трансформирующего фактора роста (TGFb-R1) в фибробластах при гипертонической болезни (А) и при ишемической болезни сердца (Б). Интенсивность реакции 2 балла (А). Иммуногистохимический метод с пероксидазной меткой.
Литература
- Guo Xia, Chen Shi-You. Transforming growth factor-βand smooth muscle differentiation // World J Biol Chem. V.26 (3).P.41-52.
- Deng H.B., Jiang C.Q., Tomlinson B. et al. A polymorphism in transforming growth factor-β1 is associated with carotid plaques and increased carotid intima-media thickness in older Chinese men: the Guangzhou Biobank Cohort Study-Cardiovascular Disease Subcohort // 2011. V.214. P.391-396.
- Rao M., Guo D., Jaber B.L.,et al. Transforming growth factor-beta 1 gene polymorphisms and cardiovascular disease in hemodialysis patients // Kidney Int. 2004. V. 66. P. 419-427.
- Saltis J., Agrotis A., Bobik A. Regulation and interactions of transforming growth factor-beta with cardiovascular cells: implications for development and disease // Clin Exp Pharmacol Physiol. 1996. V.23. P. 193-200.
- Holweg C.T., Baan C.C., Niesters H.G. et al. TGF-beta1 gene polymorphisms in patients with end-stage heart failure // J Heart Lung Transplant. 2001. V.20. P. 979–984.
- Rosenkranz S. TGF-beta 1 and angiotensin networking in cardiac remodeling // Cardiovasc Res. 2004. V.63 (3). P. 423-432.
- Hein S., Arnon E., Kostin S. et al. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms // Circulation. 2003. V. 107(7). P. 984-991.
- Villar A.V., Cobo M., Llano M. et al. Plasma levels of transforming growth factor-beta1 reflect left ventricular remodeling in aortic stenosis // PLoS One. 2009. V. 4:e8476.
- Moustakas A., Heldin C.H. The regulation of TGFbeta signal transduction // Development. 2009. V. 136. P. 3699–3714.
- Zacchigna L., Vecchione C., Notte A. et al. Emilin1 links TGF-beta maturation to blood pressure homeostasis // Cell. 2006. V. 124. P. 929–942.
- Li R.K., Li G., Mickle D.A. et al. Overexpression of transforming growth factor-beta1 and insulin-like growth factor-I in patients with idiopathic hypertrophic cardiomyopathy // Circulation. 1997. V. 96. P. 874–881.
- Pauschinger M., Knopf D., Petschauer S. et al. Dilated cardiomyopathy is associated with significant changes in collagen type I/III ratio // Circulation. 1999. V.99. P. 2750–2756.
- Sanford L.P., Kallapur S., Ormsby I., Doetschman T. Influence of genetic background on knockout mouse phenotypes // Methods Mol Biol. 2001. V. 158. P. 217–225.
- Doetschman T. Interpretation of phenotype in genetically engineered mice // Lab Anim Sci. 1999. V. 49. P. 137–143.
- Doetschman T., Barnett J., Runyan B. Transforming growth factor beta signaling in adult cardiovascular diseases and repair // Cell Tissue Res. 2012. V. 347(1). P. 203–223.
- Vincent F., Vellucci and Michael Reiss. Cloning and Genomic Organization of the Human Transforming Growth Factor-b Type I Receptor Gene // GENOMICS 1997. V. P. 278–283.
- Mathhew C.C. The isolation of high molecular weight eucariotic DNA. Methods in Molecular Biology. N.Y.: Human Press, 1984. V. 2. P. 31-34.
- Orita M., Jmahana H., Kanazawa H. et.al. Detection of polymorphism of human DNA by gel electrophoresis as single cell conformation polymorphism // Proc. Natl. Acad. Sci. 1989. V. 86. P. 2766-2770.
- Chen Т., Jackson C.R., Link А. et al. Int7G24A Variant of Transforming Growth Factor-B Receptor Type I Is Associated with Invasive Breast Cancer // Clin Cancer Res. 2006. V. 12(2). Р. 392-397.
- Stheneur C., Collod-Béroud G., Faivre L.et al.Identificationof 23 TGFBR2 and 6 TGFBR1 genemutationsandgenotype-phenotypeinvestigationsin 457 patientswithMarfansyndrometypeIandII, Loeys-Dietzsyndromeandrelateddisorders // HumMutat. 2008. V. 29(11). E284-95. doi: 10.1002/humu.20871.
- Liu X., Shan Y., Xue B. Int7G24A polymorphism (rs334354) and cancer risk // Acrch. Med. Sci. 2013. V.9(1). P. 3-7.
- Xin Jiang, Zeng He-song, Guo Yi et al. The expression of matrix metalloproteinases-9, transforming growth factor-b1and transforming growth factor-b receptor in human atherosclerotic plaque and their relationship with plague stability // Chinese Medical Journal. 2004. V. 117 (12). P. 1825-09.
- Jachec W., Foremny A., Domal-Kwiatkowska D. et al. Expression of TGF-beta1 and its receptor genes (Tbeta RI, Tbeta RII, TbetaRIII-betaglycan) in peripheral blood leucocytes in patients with idiopathic pulmonary arterial hypertension and Eisenmenger,s syndrome // Int J Mol Med. 2008. V. 21(1). P. 99-107.
- Li G., Li R.K., Mickle D.A. et.а Elevated overpression of insulin-like growth factor and transforming growth factor in the myocardium of patient with hypertrophic Thr144-9.
- Li G., Borger M.A., Williams W.G., Weisel R.D., et al. Regional overpression of insulin-like growth factor and transforming growth factor in the myocardium of patient with hypertrophic obstructive cardiomyopathy // J. Thorac Cardiovasc. Surg. 2002. V. 123. P.89-95.
- Ellmers, NicolaJ. A. Scott, SatyanarayanaMedicherla, AnnaP. Pilbrow, etal. Transforming Growth Factor-β blockade down-regulates the renin-angiotensin system and modified cardiac remodeling after myocardial infarction // The Journal of Clinical Endocrinology & Metabolism. 2008. V.149. Issue 11. P.5828-34.