QUANTIFICATION OF GENOME-WIDE DNA METHYLATION WITH METHYLATION SENSITIVE RESTRICTION-IMAGEJ ASSAY (MSR-IA)

Сучкова И.О.1, Дергачева Н.И.2, Павлинова Л.И.3, Сасина Л.К.4, Баранова Т.В.5, Паткин Е.Л.6
1 ORCID: 0000-0003-2127-0459, кандидат биологических наук, ФГБНУ «Институт экспериментальной медицины»; 2 ORCID: 0000-0002-1643-9558, студент-магистрант, РГПУ им. А.И. Герцена, ФГБНУ «Институт экспериментальной медицины»; 3 ORCID: 0000-0001-6148-1256, кандидат биологических наук, ФГБУН Институт физиологии им.И.П. Павлова, ФГБНУ «Институт экспериментальной медицины»; 4 ORCID: 0000-0002-5848-5544, кандидат биологических наук, ФГБНУ «Институт экспериментальной медицины»; 5 кандидат биологических наук, ФГБНУ «Институт экспериментальной медицины»; 6 ORCID: 0000-0002-6292-4167, профессор, доктор биологических наук, ФГБНУ «Институт экспериментальной медицины»
Работа выполнена при поддержке гранта РФФИ №15-04-04642-а
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ПОЛНОГЕНОМНОГО МЕТИЛИРОВАНИЯ ДНК С ПОМОЩЬЮ МЕТИЛ-ЧУВСТВИТЕЛЬНОЙ РЕСТРИКЦИИ И IMAGEJ АНАЛИЗА (MSR-IA)
Аннотация
В статье подробно описан метод количественного определения относительного уровня полногеномного метилирования ДНК в CCGG сайтах с помощью метил-чувствительной рестрикции с использованием программного обеспечения ImageJ для анализа электрофореграмм (Methylation Sensitive Restriction-ImageJ Assay, MSR-IA). Показано, что для определения истинного уровня полногеномного метилирования ДНК в методических подходах, в основе которых лежит использование метил-чувствительных эндонуклеаз рестрикции, необходимо использовать стандартные образцы с известным уровнем метилирования ДНК и вычислении коэффициентов уравнения линейной регрессии. Предлагаемый методический подход позволяет быстро оценить относительный уровень полногеномного метилирования ДНК и может быть использован для выявления эпигеномных нарушений, вызванных вредными факторами окружающей среды, а также в диагностических целях в клинической медицине.
Ключевые слова: метилирование ДНК, эндонуклеазы рестрикции, электрофорез, ImageJ.
Suchkova I.O.1, Dergacheva N.I.2, Pavlinova L.I.3, Sasina L.K.4, Baranova T.V.5, Patkin E.L.6
1 ORCID: 0000-0003-2127-0459, PhD in Biology, FSBSI "Institute of Experimental Medicine"; 2 ORCID: 0000-0002-1643-9558, Master student, Herzen University, FSBSI "Institute of Experimental Medicine"; 3 ORCID: 0000-0001-6148-1256, PhD in Biology, FSBIS Pavlov Institute of Physiology, FSBSI "Institute of Experimental Medicine"; 4 ORCID: 0000-0002-5848-5544, PhD in Biology, FSBSI "Institute of Experimental Medicine"; 5 PhD in Biology, FSBSI "Institute of Experimental Medicine"; 6 ORCID: 0000-0002-6292-4167, PhD in Biology, Professor, FSBSI "Institute of Experimental Medicine".
This work was supported by the Russian Foundation for Basic Research (project nos. 15-04-04642-а)
QUANTIFICATION OF GENOME-WIDE DNA METHYLATION WITH METHYLATION SENSITIVE RESTRICTION-IMAGEJ ASSAY (MSR-IA)
Abstract
The article describes the method of quantitative determining of the relative level of genome-wide DNA methylation in CCGG sites using methyl-sensitive restriction and ImageJ software for electrophoregram analysis (Methylation Sensitive Restriction-ImageJ Assay, MSR-IA). It is shown that determination of true level of genome-wide DNA methylation in approaches, which are based on the use of methyl-sensitive restriction enzymes, it is necessary to use standard samples with known level of DNA methylation and calculation coefficients of linear regression. The suggested methodical approach allows quick assessment of relative level of genome-wide DNA methylation. It can be used to identify epigenomic disturbances caused by adverse environmental factors, as well as for diagnostic purposes in clinical medicine.
Keywords: DNA methylation, restriction endonucleases, electrophoresis, ImageJ.
Введение.
Метилирование ДНК является одним из основных эпигенетических механизмов регуляции экспрессии генов, инактивации Х-хромосомы и геномного импринтинга [1-4]. Аномальное метилирование ДНК может приводить к отклонениям в эмбриогенезе и развитию различных патологий во взрослом состоянии, причем, если нарушения паттерна метилирования ДНК произошли в половых клетках, то такой измененный статус метилирования может наследоваться [3-6]. Причинами нарушений метилирования ДНК могут быть как эндогенные, так и экзогенные факторы. В связи с этим во всем мире активно проводятся исследования метилирования ДНК не только при различных заболеваниях человека, но и при воздействии вредных факторов окружающей среды [7].
Для оценки уровня полногеномного, сайт- и ген(локус)-специфического метилирования ДНК предложен широкий спектр методов, описанию которых посвящено много обзоров литературы [8-18]. Все методы анализа метилирования ДНК базируются на трех основных принципах и их комбинациях (ферментативном гидролизе ДНК метил-чувствительными рестриктазами, химической модификации метилированного цитозина (бисульфитная конверсия), взаимодействии антител или метил-специфичных белков с метилированным цитозином) в сочетании с такими техническими подходами как хроматография, электрофорез, полимеразная цепная реакция, секвенирование, иммунопреципитация, масс-спектрометрия, ДНК-микрочип технология. Каждый из используемых в настоящее время методов имеет свои ограничения и недостатки, поэтому остается актуальным разработка и оптимизация методов анализа метилирования ДНК, которые одновременно были бы достаточно просты в применении, экономичны и эффективны. Цель данной работы состояла в оптимизации метода метил-чувствительной рестрикции с использованием ImageJ анализа электрофореграмм (Methylation Sensitive Restriction-ImageJ Assay, MSR-IA) для количественной оценки полногеномного метилирования ДНК в CCGG сайтах.
Материалы и методы.
В представленной работе в качестве стандартных образцов с известным уровнем полногеномного метилирования ДНК были использованы образцы коммерческой неметилированной и метилированной ДНК человека («Human methylated and non-methylated DNA Set», «Zymo Research»). Неметилированная форма ДНК получена из клеточной линии HCT116 DKO, которая является генетическим нокаутом по двум ДНК метилтрансферазам (DNMT1 и DNMT3b). ДНК этих клеток имеет низкий уровень метилирования (< 5% CpG) и поэтому часто используется как негативный контроль при анализе метилирования CpG динуклеотидов. Метилированная форма ДНК (100% CpG) этой же клеточной линии получена фирмой-производителем in vitro с помощью CpG-метилазы и служит в качестве позитивного контроля при анализе метилирования CpG динуклеотидов. На основе неметилированной и метилированной ДНК был сделан стандартный образец, моделирующий 50% уровень метилирования ДНК.
В качестве образца для анализа была использована ДНК с неизвестным уровнем метилирования, которая была выделена методом безфенольной хлороформной экстракции из лейкоцитов периферической венозной крови женщины 38 лет. Забор крови проводили в коммерческие вакуумные пробирки с K-ЭДТА (можно с Li-гепарином, как показывает наш опыт, на качество получаемой ДНК это не влияет). Цельную кровь инкубировали при 370С в течение 30-60 мин. Плазму с лейкоцитами переносили в 2 мл пробирки. Лейкоциты осаждали центрифугированием при 1000g в течение 10 мин. Если в осадке лейкоцитов присутствовала примесь эритроцитов, то проводили дополнительную инкубацию клеток в 1 мл раствора 5мМ ЭДТА при комнатной температуре в течение 20-30 минут. Затем полученный осадок лейкоцитов инкубировали в течение ночи при 550С в буфере для выделения ДНК (50 mM TrisHCl pH 8.0; 10 mM ЭДТА pH 8.0; 100 mM NaCl, 1% SDS, 1 мкг/мкл протеинкиназа К). Экстракцию ДНК проводили с помощью смеси хлороформ:изоамиловый спирт (24:1) (без стадии с фенолом и повторяли 3 раза). Центрифугирование проводили при 1000g в течение 10 мин. ДНК осаждали этанолом. Высушенный осадок ДНК растворяли в бидистиллированной воде. Концентрацию полученной ДНК определяли спектрофотометрически.
Ферментативный гидролиз ДНК проводили с помощью метил-чувствительных эндонуклеаз рестрикции MspI и HpaII, следуя рекомендациям фирмы-производителя ферментов («Thermo Scientific»). Рестрикционная смесь объемом 15 мкл содержала 33 мМ Трис-ацетат (pH 7.9), 10 мМ ацетат магния, 66 мМ ацетат калия, 0.1 мг/мл BSA, ферменты MspI либо HpaII по 10 ед. на 350 нг геномной ДНК. Смесь инкубировали при 370С в течение 5.5 часов. Для одного и того же исследуемого образца ДНК в опыт брали три варианта: с MspI, с HpaII и без добавления ферментов. Последний вариант служил контролем сохранности ДНК в реакционном буфере.
В качестве дополнительного контроля эффективности ферментативного гидролиза использовали плазмидную ДНК pUC19 (GenBank accession number L09137) («СибЭнзим»). pUC19 (2686 пн) имеет G+C состав 50.6%, содержит 173 CpG динуклеотидов (6.4%), 13 из которых находятся в сайте узнавания CCGG для MspI (HpaII) (0.5%). Для сравнения геномная ДНК человека имеет G+C состав 41-42%, частота CpG только около 1% [19, 20]. Теоретические расчеты показывают, что вероятность встречаемости CCGG составляет 44, т.е. через каждые 256 нуклеотидов, следовательно, в диплоидной клетке человека (5.8х109пн) может содержаться около 2.2х107 CCGG сайтов (0.4%). Поскольку 1 пг ДНК это 0.965x109 пн [21], то в 100 нг геномной ДНК человека (17.2х103 клеток) теоретически должно быть примерно 3.8х1011 CCGG сайтов, а в 100 нг pUC19 (35.9х109 молекул) около 4.7х1011 CCGG. Поэтому эффективность ферментативного гидролиза плазмидной ДНК pUC19, взятой в реакцию в большем количестве (500 нг), чем геномная ДНК (350 нг), может служить адекватным показателем полноты рестрикции исследуемой геномной ДНК человека.
После ферментативного гидролиза ДНК проводили электрофорез в 1% агарозном геле на 0.5хТВЕ (50 минут, 40 мА). В лунки наносили весь объем рестрикционной смеси, к которому было добавлено по 2 мкл буфера для нанесения (0.01% бромфеноловый синий, 30% глицерин). ДНК визуализировали окрашиванием бромистым этидием (1мкг/мл) в течение 15 минут. Гель фотографировали (рис.1).
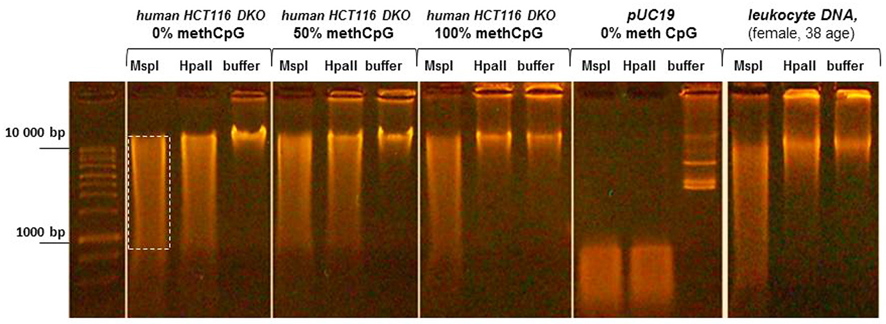
Рис. 1. Электрофореграмма ДНК человека и плазмиды pUC19 после рестрикции MspI и HpaII (1% агарозный гель). Пунктирным прямоугольником приведен пример области измерения в ImageJ интенсивности флюоресценции ДНК, окрашенной бромистым этидием.
Для оценки относительного уровня полногеномного метилирования ДНК изображения электрофореграмм в формате jpg обрабатывались программным комплексом ImageJ [22]. Для одного и того же исследуемого образца измеряли интенсивность флюоресценции Mean gray value («MGV») шлейфа фрагментированной ДНК на дорожках после рестрикции MspI и HpaII. Площадь измерений для MspI и HpaII дорожек была одинаковая и включала в себя всю ширину дорожки, начиная с зоны фрагментации геномной ДНК до области нахождения краски для нанесения бромфенолового синего (рис. 1). Значения «MGV» для "MspI-вариантов" делили на значения «MGV» для "HpaII-вариантов" так получали значение «Relative gray value» («RGV») относительного уровня полногеномного метилирования ДНК по CCGG сайтам, выраженное в условных единицах (c.u.). Стандартная ошибка среднего вычислялась по формуле SD/√N, где SD-стандарное отклонение, N – количество пикселей в области измерений.
На основании значений «RGV», полученных для стандартных образцов ДНК с известным уровнем метилирования, в программе Excel вычисляли коэффициенты уравнения линейной регрессии и строили стандартную прямую зависимости уровня флюоресценции ДНК от уровня метилирования ДНК. Затем для образца с неизвестным уровнем метилирования ДНК устанавливали относительный уровень полногеномного метилирования CCGG сайтов, выраженный в %, используя полученные коэффициенты уравнения линейной регрессии (y=a+bX). Стандартная ошибка вычислялась по формуле σ /√N, где N - количество образцов, использованных при построении уравнения линейной регрессии; σ-погрешность предсказания формулы. Значения σ вычислялись по формуле σ = σy√(1-R2), где σy2 - оценка дисперсии случайной величины Y (т.е. в нашем случае % CCmeGG); R – коэффициент корреляции [23].
Результаты и обсуждение.
Рассматриваемый в работе метод основан на классическом подходе с использованием метил-чувствительных эндонуклеаз-изошизомеров MspI и HpaII [24]. Оба фермента распознают сайт 5’-CCGG-3’, но не активны, если был метилирован внешний цитозин. В тоже время при наличии метилирования внутреннего цитозина 5’-CCmeGG-3’ рестриктаза HpaII не распознает сайт, но всё же может его разрезать в случае гемиметилированного состояния, но с меньшей эффективностью. Тогда как MspI разрезает этот сайт независимо от состояния метилирования внутреннего цитозина [25, 26].
В данной работе, используя коммерческие образцы с известным уровнем метилирования ДНК (линии HCT116 DKO человека), была проверена эффективность применения нескольких формул, описанных в публикациях [8, 26, 27], для определения относительного уровня полногеномного метилирования CCGG сайтов в сравнении с вариантом вычислений, предлагаемых нами (см. «Материалы и методы», Таблица 1). Вычисления проводились по следующим формулам:
Формула № 1 [8]:  ,
,
Формула № 2 [26]: ,
Формула № 3 [27]: 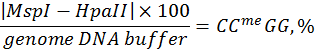 ,
,
где MspI и HpaII – флюоресценция образца после рестрикции соответствующей рестриктазой.
Таблица 1. Относительный уровень полногеномнгого метилирования CCGG сайтов в коммерческих образцах с известным уровнем метилирования ДНК (линия HCT116 DKO человека), вычисленный по формулам, описанным в работах [8, 26, 27].
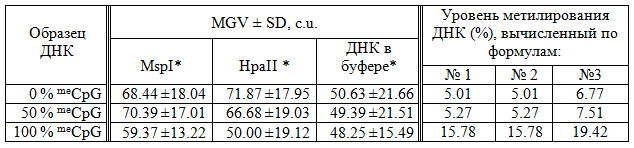
Примечание: * - уровень флюоресценции ДНК в агарозмом геле по результатам измерений в ImageJ; MGV-mean gray value; SD-standard deviation; c.u.- conventional units
Как видно из таблицы, получаемые значения относительного уровня полногеномного метилирования ДНК с использованием вышеописанных формул не дают точную информацию об истинном уровне метилирования ДНК, поэтому нами был использован другой вариант вычислений, как описано в разделе «Материалы и методы». Значения коэффициентов уравнения линейной регрессии y=423.668x-401.206 были получены с помощью регрессионного анализа в программе Excel и online-версии «Математическая статистика. Ковариация. Корреляция. Линейная регрессии» [23]. Коэффициент корреляции R=0.998, уровень значимости р=0.042, погрешность уравнения σy/x=2.702, стандартная ошибка=1.560. В нашем случае Х (ось ОХ) – это значения «Relative gray value» («RGV») для стандартных образцов с известным уровнем метилирования ДНК; Y (ось OY) – это значения относительного уровня метилирования ДНК, выраженные в % (рис. 2). Результаты, представленные в таблице 2, показывают, что вычисленные таким способом значения уровня полногеномного метилирования ДНК, выраженные в %, более соответствуют действительности, чем при вычислении по формулам из публикаций [9, 26, 27].
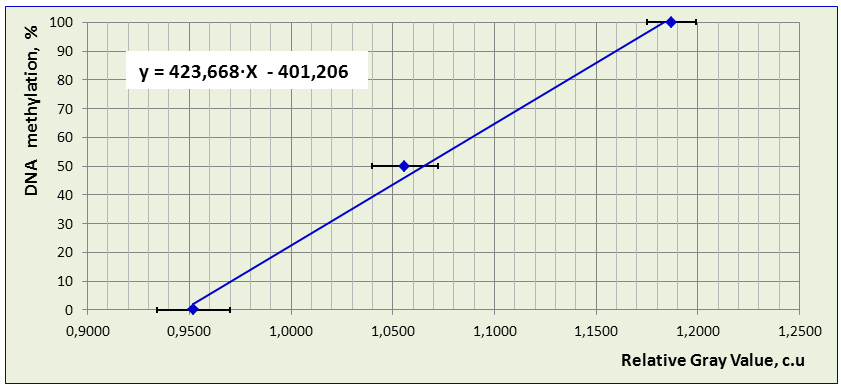
Рис. 2. Графическое изображение уравнения линейной регрессии, полученного для стандартных образцов ДНК клеточной линии человека HCT116 DKO c известным уровнем метилирования ДНК. График отражает прямую линейную зависимость между полученными в ImageJ значениями «Relative Gray Value» и уровнем полногеномного метилирования ДНК в CCGG сайтах.
Таблица 2. Относительный уровень полногеномного метилирования CCGG сайтов в коммерческих образцах с известным уровнем метилирования ДНК (линия HCT116 DKO человека) по результатам измерений в ImageJ и использованием коэффициентов уравнения линейной регрессии, вычисленных в Excel.
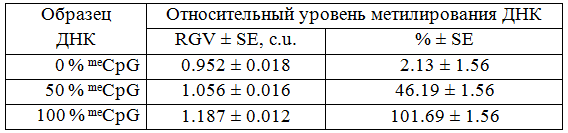
Примечание: RGV-relative gray value (MspI/HpaII); SE-standard error; c.u.- conventional units
Следует отметить, что для некоторых направлений научных исследований, где не требуется установления точного уровня метилирования ДНК (например, при изучении влияния ксенобиотиков на эпигеном), достаточно вычислить значения «RGV». В этом случае, чем больше будет значения «RGV» (выраженное в условных единицах), тем более метилированной является ДНК (таблица 2).
По аналогичной схеме был проведен анализ полногеномного метилирования ДНК человека с неизвестным уровнем метилирования (женщина, 38 лет) (рис.1, 3). Для данного образца ферментативный гидролиз проводили с разными концентрациями ДНК (400, 200, 100 нг), каждый вариант был в трех экземплярах для доказательства воспроизводимости результатов.

Рис. 3. Уровень полногеномного метилирования CCGG сайтов в ДНК лейкоцитов женщины. Представлены количественные оценки метилирования ДНК, выраженные в условных единицах (А) и процентах (В). На примере образцов ДНК разных концентраций (каждый вариант был сделан в трех экземплярах).
Показано, что вычисленные значения относительного уровня метилирования ДНК не зависят от концентрации исходной ДНК, взятой в анализ (Таблица 3). Необходимо подчеркнуть, что при использовании метода MSR-IA не следует брать в ферментативный гидролиз низкие концентрации ДНК (менее 150 нг), т.к. в этом случае повышается вероятность ошибочного установления относительного уровня метилирования ДНК, выраженного в процентах (Таблица 3).
Таблица 3. Относительный уровень полногеномного метилирвания CCGG сайтов в ДНК лейкоцитов человека. Полученные значения представлены в условных единицах и в процентах.
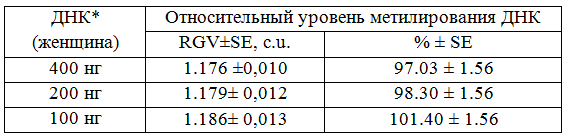
Примечание: * - средние значения по 3 экземплярам-дублям для каждого варианта концентраций ДНК; RGV-relative gray value; SE-standard error; c.u.- conventional units
Следует отметить, что по одним данным доля метилированного цитозина в геноме человека в норме составляет 80-90 % [9], по другим – 70-80% [13], но эти значения достаточно усредненные и не учитывают возможные индивидуальные различия, а также дифференциальный ткане/клетка-специфичный и локус-специфичный характер метилирования ДНК, поэтому полученный нами уровень полногеномного метилирования CCGG в лейкоцитах женщины 38 лет вписывается в эти рамки условной нормы.
Заключение. Полученные в данной работе данные говорят о необходимости использования стандартных образцов с известным уровнем метилирования ДНК и необходимости вычисления коэффициентов уравнения линейной регрессии для определения истинного уровня полногеномного метилирования ДНК, выраженного в процентах. Результаты исследования указывают на то, что метод MSR-IA может быть использован для определения уровня полногеномного метилирования ДНК, хотя, он имеет определенные ограничения. К недостаткам метода относится то, что полногеномное метилирование анализируется только по сайтам узнавания метил-чувствительных рестриктаз и для исследования одного образца требуется порядка 1 мкг ДНК. Кроме того, всегда существует вероятность неполного гидролиза ДНК или различий в активности ферментов, чувствительных и нечувствительных к метилированию. Для исключения этого в эксперимент рекомендуется брать контрольный образец плазмидной ДНК (например, pUC19, pBR322, pBlueScript) в концентрациях, превышающих концентрации геномную ДНК человека, взятой в анализ. Важным преимуществом метода MSR-IA является методическая простота, меньшая трудоемкость и достаточная дешевизна по сравнению с методами, требующими использования антител, метил-специфичных белков, бисульфитной обработки, ПЦР, секвенирования и т.д. Предлагаемый методический подход может быть использован для выявления эпигеномных нарушений, вызванных вредными факторами окружающей среды, а также в диагностических целях в клинической медицине.
Литература (References)
- Bird, A. DNA methylation patterns and epigenetic memory // Genes and Development. – – Vol. 16. – P. 6-21.
- Patkin, E.L. Epigenetic mechanisms for primary differentiation in mammalian embryos // Rev. Cytology. – 2002. – Vol. 216. – P.81-130.
- Lim, D.H.K., Maher, E.R. DNA methylation: a form of epigenetic control of gene expression // Obstetrician and Gynaecologist. – 2010. – Vol. 12. – P. 37-42.
- Smith, Z.D., Meissner, A. DNA methylation: roles in mammalian development // Rev. Genetics. – 2013. – Vol. 14. – P. 204-220.
- Patkin, E.L. Epigenetical mechanisms of human common diseases. – St.Petersburg: Nestor-Story, – 200 p. (Паткин Е.Л. Эпигенетические механизмы распространенных заболеваний человека. – Санкт-Петербург: Нестор-История, 2008. – 200 с.)
- Patkin, E.L., Quinn, J. Epigenetical mechanisms of susceptibility to complex human diseases // J. Genetics: Applied Res. – 2011. – Vol. 1 (5). –P. 436-447.
- Patkin, E.L., Sofronov, G.A. Population epigenetics. ecotoxicology. and human diseases // J. Genetics: Applied Res. – 2013. – Vol. 3 (5). – P. 338-351.
- Filina, Yu.V., Gabdulkhakova, A.G.. Arleyevskaya, M.I. The methods of analysis of DNA methylation // Rus. Clinical laboratory diagnostics. – – №8. – P. 15-18. (Филина Ю.В., Габдулхакова А.Г., Арлеевская М.И. Методы анализа метилирования ДНК // Клиническая лабораторная диагностика. – 2012. – № 8. – P. 15-18.)
- Skryabin, N.A., Kashevarova, A.A., Denisov, E.V., Lebedev, I.N. Methods of DNA methylation analysis: possibilities and limitations of their application in oncology // Siberian oncological journal. – – № 6 (60). P. 64-69. (Скрябин Н.А., Кашеварова А.А., Денисов Е.В., Лебедев И.Н. Методы исследования метилирования ДНК: возможности и перспективы использования в онкологии // Сибирский онкологический журнал. – 2013. – № 6 (60). – P. 64-69.)
- Oakeley, E.J. DNA methylation analysis: a review of current methodologies // Pharmacology and Therapeutics. – 1999. – Vol. 84. – P. 389-400.
- Zuo, T., Tycko, B., Liu, T-M., Lin, H-J.L., Huang, T.H-M. Methods in DNA methylation profiling // – 2009. – Vol. 1(2). – P. 331-345.
- Shen, L. A., Waterland, R.A. Methods of DNA methylation analysis // Cur. Opin. Clin. Nutrition and Metabolic Care. – 2007. – Vol. 10. – P. 576-581.
- Zilberman, D., Henikoff, S. Genome-wide analysis of DNA methylation patterns // Development. – 2007. – Vol. 134. – P. 3959-3965.
- Gupta, R., Nagarajan, A., Wajapeyee, N. Advances in genome-wide DNA methylation analysis // Biotechniques. – – Vol. 49(4).- P. iii–xi.
- Laird, P.W. Principles and challenges of genome-wide DNA methylation analysis // Nat. Rev. – 2010. – Vol. 11. – P. 191-203.
- Rauch, T.A., Pfeifer, G.P. Methods for assessing genome-wide DNA methylation. – Handbook of epigenetics: The new molecular and medical genetics. – Elsevier Inc. – – Chapter 9. – P. 135-147.
- Dhingra, T., Mittal, K., Sarma, G.S. Analytical Techniques for DNA Methylation - An Overview // Pharm. Analysis. – 2014. – Vol. 10. – P. 71-85.
- Wang, S., Lv, J., Zhang, L., Dou, J., et al. MethylRAD: a simple and scalable method for genome-wide DNA methylation profiling using methylation-dependent restriction enzymes // Open Biology. – 2015. – Vol. 5. – P. 150130.
- Scarano, E., Iaccarino, M., Grippo, P., Parisi, E. The heterogeneity of thymine methyl group origin in DNA pyrimidine isostichs of developing sea urchin embryos // Natl. Acad. Sci. USA. – 1967. – Vol. 57 (5). – P. 1394–1400.
- Kryukov, K. Genome Composition Database. List of species. Saitou lab. National Institute of Genetics. (2006-2010) [Electronic resource] URL: http://esper.lab.nig.ac.jp/study/genome/?page=genome_composition_database_species_list (Accessed 25.03.2016.)
- Lewin, B. Genes. J. Wiley and Sons. New York. Chichester. Brisbane. Toronto. Singapure. (Льюин Б. Гены. Пер с англ. – М.: Мир, 1987. – 544 с.)
- Rasband, W.S. ImageJ. U.S. National Institutes of Health. Bethesda. Maryland. USA. (1997-2015). [Electronic resource] URL: http://imagej.nih.gov/ij/ (Accessed 01.09.2014.)
- OnLine-higher mathematics services. Math statistics. Covariance, correlation, linear regression. [Electronic resource] URL: http://www.math-pr.com/stst_3v_1.php (Accessed 12.03.2016.)
- Waalwijk, C., Flavell, R.A. MspI an isoschizomer of HpaII which cleaves both unmethylated and methylated HpaII sites // Acids Res. – 1978. – Vol. 5. – P. 3231-3236.
- New England Biolabs Catalog and Technical Reference. New England Biolabs. Ipswich. MA. 2005-2006. – P. 266–267.
- Wentzel, J.F., Gouws, C., Huysamen, C., van Dyk, E., Koekemoer G., Pretorius P.J. Assessing the DNA methylation status of single cells with the comet assay // Analytical Biochemistry. – 2010. – Vol. 400. – P. 190-194.
- Errante, P.R., Perazzio, S.F., Rodrigues, F.S.M., Ferraz, R.R.N., Caricati-Neto, A. Global DNA methylation pattern and correlation with complement system proteins in Brazilian patients with systemic lupus erythematosus // ConScientiae Saúde. – 2014. – Vol. 13(4). – P. 516-523.