КВАНТОВОХИМИЧЕСКОЕ ИССЛЕДОВАНИЕ ЭНЕРГЕТИКИ ВОДОРОДНОЙ СВЯЗИ И ПРОСТРАНСТВЕННОЙ СТРУКТУРЫ КОНФОРМАЦИЙ ТИОПРОИЗВОДНЫХ АЦЕТИЛАЦЕТОНА
Иванов Ю.В.1, Жамская Н.Н.2, Бянкина Л.С.3, Апанасенко О.А.4, Каткова С.А.5
1ORCID: 0000-0001-6846-892, Доцент, Кандидат химических наук, 2,3,4,5Доцент, Кандидат химических наук, Дальневосточный государственный технический рыбохозяйственный университет
КВАНТОВОХИМИЧЕСКОЕ ИССЛЕДОВАНИЕ ЭНЕРГЕТИКИ ВОДОРОДНОЙ СВЯЗИ И ПРОСТРАНСТВЕННОЙ СТРУКТУРЫ КОНФОРМАЦИЙ ТИОПРОИЗВОДНЫХ АЦЕТИЛАЦЕТОНА
Аннотация
С целью исследования энергии водородной связи а также изучения влияния расчетных базисов в рамках неэмпирического подхода были рассчитаны структуры конформаций енолизованных форм тиоацетилацетона в одноэлектронных методах, а также с учетом электронной корреляции методами возмущения Меллера-Плессета второго порядка (MP2) и функционала электронной плотности (DFT). Показано, что учёт эффекта электронной корреляции и расширение базиса для одноэлектронного метода может существенно влиять на качественную картину энергии конформеров и таутомеров.
Ключевые слова: тиоацетилацетон, водородная связь, конформация, неэмпирические расчёты, пространственное строение, энергия корреляции.
Ivanov Yu.V.1, Zhamskaya N.N.2, Biankina L.S.3, Apanasenko O.A.4, Katkova S.A.5
1ORCID: 0000-0001-6846-892, Associate Professor, PhD in Chemistry, Far Eastern State Technical Fisheries University, 2,3,4,5Associate Professor, PhD in Chemistry, Far Eastern State Technical Fisheries University
QUANTUM CHEMICAL STUDY OF HYDROGEN BOND AND SPATIAL STRUCTURE OF CONFORMATIONS OF THIO-DERIVATIVES OF ACETYLACETONE
Abstract
In order to study the energy of the hydrogen bond and the effect of the calculated bases in the framework of the non-empirical approach, we estimated the conformational structures of the enolisated forms of thioacetylacetone in one-electron methods taking into account the electron correlation by the methods of the Möller-Plesset perturbation of the second order (MP2) and the electron density functional (DFT). It is shown that the electron correlation effect and the expansion of the basis for the one-electron method can significantly affect the qualitative picture of the energy of conformers and tautomers.
Keywords: thioacetylacetone, hydrogen bond, conformation, non-empirical calculations, spatial structure, correlation energy.
Ранее неоднократно изучались строение, конформационные переходы таутомеров 1,3-дикетонов и их производных различными экспериментальными и теоретическими квантовохимическими полуэмпирическими и неэмпирическими методами [1], [2], [3]. Между тем, наличие такого атома третьего периода, как сера, может оказывать существенное влияние как на сами свойства таутомеров 1,3-тиокарбонильного соединения, так и на процесс таутомеризации в целом. В частности, помимо енольных форм (V, VI, VII), возможна также енолизация исходного тиодикетона I по атому серы, с образованием тиольных таутомеров II, III, IV (рис. 1).
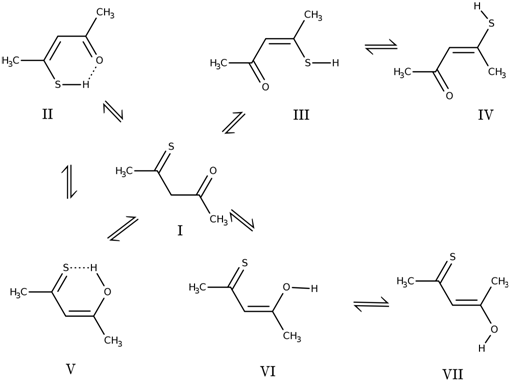
Рис. 1 – Конформации и таутомерные формы тиоацетилацетона
Вследствие сильной подвижности атома водорода точное измерение различными экспериментальными методами сталкивается с определенными трудностями [3], что делает еще более актуальной задачу теоретического изучения подобного плана тиокарбонильных соединений.
В настоящей работе ставилась задача исследования неэмпирическими методами квантовой химии геометрического строения конформеров тиоацетилацетона, как простейшего представителя 1,3-тиокарбонильного соединения, связь конформационных форм его таутомеров с энергией водородной связи, а также изучалось влияние расчетного базиса и полученной в ходе различных приближений энергии электронной корреляции на расчетную величину энергии молекулы и ее пространственное строение. Все расчеты проводились в корреляционно-согласованном базисе cc-pVDZ, и улучшенном aug-cc-pVDZ. Для учета корреляционных поправок использовался метод возмущений Меллера-Плессета второго порядка (MP2) и метод функционала электронной плотности (DFT) с применением гибридного функционала B3LYP. Для расчетов использовалась программа MPQC [4] гибридного функционала B3LYP. Для расчетов использовалась программа MPQC [4]
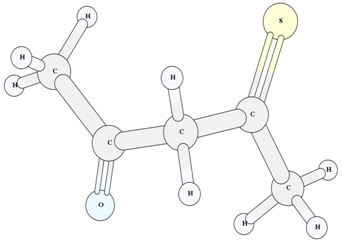
Рис. 2 – Структура исходного тиокетона I
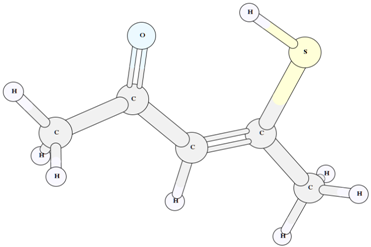
Рис. 3 – Структура енольного таутомера II
Структуры таутомерных молекул и их конформеров оптимизированные в расчетах b3lyp/cc-pVDZ представлены на рис. 2–8. Для вывода графических моделей использовалась программа Gabedit [5].
Следует отметить, что структурные параметры молекул, полученные в различных методах различаются незначительно, однако видно (табл. 1), что расширение базиса cc-pVDZ до aug-cc-pVDZ приводит к некоторому завышению межатомных расстояний, тогда как расчеты с учетом электронной корреляции напротив приводят к их заметному сокращению.
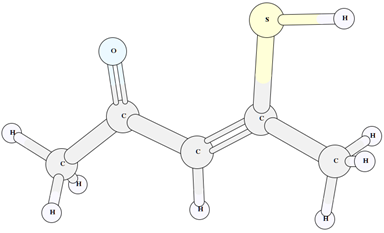
Рис. 4 – Структура енольного таутомера III
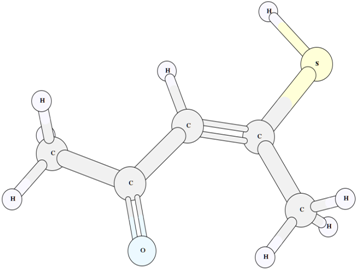
Рис. 5 – Структура енольного таутомера IV
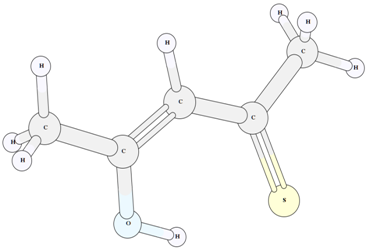
Рис. 6 – Структура енольного таутомера V
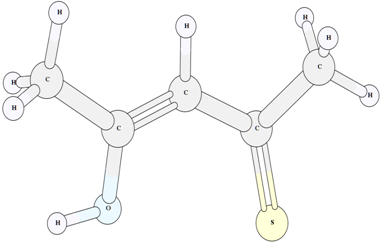
Рис. 7 – Структура енольного таутомера VI
Таблица 1 – Равновесные межатомные расстояния S—O , Å
| Базис | I | III | VI | II | V |
| cc-pVDZ | 4,328 | 2,818 | 3,076 | 3,107 | 3,000 |
| aug-cc-pVDZ | 4,364 | 2,830 | 3,082 | 3,108 | 3,005 |
| MP2/cc-pVDZ | 4,362 | 2,731 | 3,030 | 3,024 | 2,914 |
| B3lyp/cc-pVDZ | 4,361 | 2,711 | 3,050 | 2,979 | 2,909 |
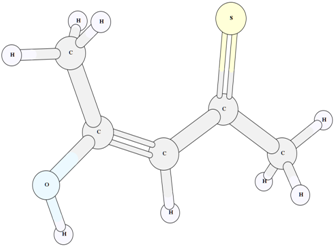
Рис. 8 – Структура енольного таутомера VII
Длина водородной связи в тех таутомерах, где она может образовываться (II и V), также обнаруживает те же тенденции при расширении базиса и учете электронной корреляции (табл. 2). Кроме того, вполне ожидаемо ее увеличение для атома серы, однако оно крайне незначительно, и не превышает 0,1 – 0,15 Å.Таблица 2 – Ддина водородной связи, Å
| Связь | cc-pVDZ | aug-cc-pVDZ | MP2/cc-pVDZ | B3lyp/cc-pVDZ |
| O--H | 2,033 | 2,041 | 1,818 | 1,732 |
| S--H | 2,136 | 2,148 | 1,963 | 1,957 |
Из таблицы 3, в которой приведены относительные энергии рассчитанных соединений, видно, что учёт корреляционных эффектов оказывает весьма существенное влияние на результат расчета энергетических характеристик молекул. Так, если расширение числа базисных функций со 142 (базис cc-pVDZ) до 237 (базис aug-cc-pVDZ) приводит к выигрышу порядка 40 – 45 кДж/моль, то энергия корреляции, полученная методом возмущений MP2, составляет более трех с половиной, а метод функционала электронной плотности показывает корреляционную энергию более шести тысяч кДж/моль. Очевидно, что величины такого масштаба не могут не оказывать влияния на качественный результат проводимых расчетов, что и наблюдается при сравнении относительной энергии таутомеров (табл. 4). В частности, одноэлектронные расчеты в базисе cc-pVDZ указывают, что енолизация по по атому серы (структура II) энергетически выгодней енолизации по кислороду (структура V). Улучшение базиса до aug-cc-pVDZ хотя качественно меняет этот результат, однако количественный энергетический выигрыш в этом случае невелик. В то же время, учет энергии корреляции приводит к явному предпочтению енолизации тиокетона по кислороду, а таутомеры VI и VII являются энергетически невыгодными. Также можно видеть, что енолизация по атому серы без образования водородной связи (структуры III и IV) также не приводит к сколько-нибудь значительному энергетическому выигрышу по сравнению с неенолизованой формой (I). К настоящему времени разработано и продолжает разрабатываться [см. напр. 7] множество методов учета электронной корреляции в рамках различных приближений, однако предпочтительность того или иного метода пока может оцениваться только на основе сравнительного анализа с экспериментальными данными.
Таблица 3 – Энергия электронной корреляции таутомерных и енольных форм тиоацетилацетона, кДж/моль
| Базис | I | II | III | IV | V | VI | VII |
| cc-pVDZ | 6250,5 | 6261,9 | 6261,2 | 6256,3 | 6281,5 | 6269,8 | 6269,8 |
| aug-cc-pVDZ | 6207,6 | 6228,8 | 6221,2 | 6218,8 | 6237,7 | 6222,0 | 6225,7 |
| MP2/cc-pVDZ | 3572,4 | 3589,1 | 3586,1 | 3587,1 | 3596,9 | 3589,2 | 3590,1 |
| B3lyp/cc-pVDZ | 0,0 | 0,0 | 0,0 | 0,0 | 0,0 | 0,0 | 0,0 |
Несмотря на то, что в виду таких особенностей ab initio расчетов, как учет многоцентровых взаимодействий не существует достаточно строгого метода, позволяющего выделить отдельно двухатомную энергию и энергию связи в частности, тем не менее, расчет в данном случае позволяет сделать приблизительную оценку энергии водородной связи, сравнивая энергию соответствующих таутомеров с водородной связью и без нее. Так, по данным DFT-расчетов конформеры II и III различаются приблизительно на 13 кДж/моль, а V и VI на более, чем 70 кДж/моль (табл. 4). Однако такой неожиданный результат может объясняться не только более высокой прочностью водородной связи S--H, но и относительно невыгодным взаимным расположением других атомов, в частности, близким расположением атомов кислорода и серы и, вследствие этого, их взаимным отталкиванием.
Таблица 4 – Относительная молекулярная энергия таутомеров тиоацетилацетона, кДж/моль
| Базис | I | II | III | IV | V | VI | VII |
| cc-pVDZ | 11,39 | 0,00 | 12,76 | 15,25 | 6,48 | 67,82 | 50,25 |
| aug-cc-pVDZ | 5,78 | 4,12 | 10,06 | 15,10 | 0,00 | 57,29 | 43,45 |
| MP2/cc-pVDZ | 11,37 | 5,25 | 15,81 | 24,21 | 0,00 | 65,22 | 48,62 |
| B3lyp/cc-pVDZ | 35,89 | 13,05 | 26,60 | 34,00 | 0,00 | 72,99 | 55,45 |
Таким образом, на основании проведенных исследований можно сделать вывод, что наиболее прочная водородная связь образуется за счет енолизации по кислороду. Расширение расчетного базиса не приводит к качественно новым результатам, тогда как использование методов, учитывающих корреляционные эффекты, приводят к взаимно непротиворечивым результатам, что и обуславливает их предпочтительность для исследования конформационных эффектов карбонильных соединений, содержащих атомы элементов III периода. Полные энергии конформеров и таутомеров тиоацетилацетона также различаются незначительно, что и объясняет легкость переходов между ними.
Список литературы / References
- Шокова Э.А. 1,3-дикетоны. Синтез и свойства / Э. А. Шокова, Дж. К. Ким, В. В. Ковалев // Журнал органической химии. – 2015. – Т. 51. – Вып. 6 – С. 773-847.
- Иванов Ю.В. Исследование таутомерии иминопроизводных оксиацетофенона неэмпирическими методами / Ю. В. Иванов // Современные проблемы химической науки и образования, Чебоксары, 2012. – 19-20 апреля. – С. 84-85.
- Кострюкова Т.С. Получение фторированных β-дикетонов / Т. С. Кострюкова, Н. В. Васильев // Вестник Московского государственного областного университета серия «Химия». – – № 1. – С. 150-156.
- The Massively Parallel Quantum Chemistry Program (MPQC), Version 2.3.1 / Curtis L. Janssen, Ida B. Nielsen, Matt L. Leininger, Edward F. Valeev, Joseph P. Kenny, Edward T. Seidl, Sandia National Laboratories, Livermore, CA, USA, 2008.
- Allouche, A.-R. Gabedit. A graphical user interface for computational chemistry softwares // Journal of Computational Chemistry. – 2011 – № 32 – 174-182.
- Аникин Н.А. Новая аппроксимация DFT для быстрых расчетов био- и наноструктур / Н. А. Аникин, М. Б. Кузьминский // Информационно-вычислительные технологии в науке – 2011, Сб. Тезисов. – М.: 2011 – С. 29-30.
Список литературы на английском языке / References in English
- Shokova Je.A. 1,3-diketony. Sintez i svojstva [1,3-diketones. Synthesis and properties] / Je. A. Shokova, Dzh. K. Kim, V. V. Kovalev // Zhurnal organicheskoj himii [The Journal of organic chemistry]. – 2015. – V. 51. – Iss. 6 – P. 773-847. [in Russian]
- Ivanov Ju.V. Issledovanie tautomerii iminoproizvodnyh oksiacetofenona nejempiricheskimi metodami [Ab initio investigation of tautomerism iminoderivatives of oxiacetophenon] / Ju. V. Ivanov // Sovremennye problemy himicheskoj nauki i obrazovanija [Modern problems of chemical science and education]. – Cheboksary, 2012. – 19-20 April. – P. 84-85. [in Russian]
- Kostrjukova T.S. Poluchenie ftorirovannyh β-diketonov [Preparation of β-diketones fluoroderivatives] / T. S. Kostrjukova, N. V. Vasil'ev // Vestnik Moskovskogo gosudarstvennogo oblastnogo universiteta serija «Himija» [Bulletin MRSU, chemistry section]. – 2012. – № 1 – P. 150–156. [in Russian]
- The Massively Parallel Quantum Chemistry Program (MPQC), Version 2.3.1, Curtis L. Janssen, Ida B. Nielsen, Matt L. Leininger, Edward F. Valeev, Joseph P. Kenny, Edward T. Seidl, Sandia National Laboratories, Livermore, CA, USA, 2008.
- Allouche, A.-R. Gabedit – A graphical user interface for computational chemistry softwares // Journal of Computational Chemistry. – 2011 – № 32 – 174-182.
- Anikin N.A. Novaja approksimacija DFT dlja bystryh raschetov bio- i nanostruktur [New DFT approximation for bio- and nanostructural calculations] / N. A. Anikin, M. B. Kuz'minskij // Informacionno-vychislitel'nye tehnologii v nauke [Informational and computational science technologies]. – 2011, Sb. Tezisov. – M.: 2011 – P. 29-30. [in Russian]