МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКАЯ ДИАГНОСТИКА ГЕПАТОЛЕНТИКУЛЯРНОЙ ДЕГЕНЕРАЦИИ: ОСОБЕННОСТИ И ПЕРСПЕКТИВЫ

МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКАЯ ДИАГНОСТИКА ГЕПАТОЛЕНТИКУЛЯРНОЙ ДЕГЕНЕРАЦИИ: ОСОБЕННОСТИ И ПЕРСПЕКТИВЫ
Обзорная статья
Гарбуз М.М.1, *, Овчинникова Е.В.2, Овчинников А.В.3, Кумейко В.В.4, Овчинникова А.А.5
1 ORCID: 0000-0003-0272-4478;
1-5 Дальневосточный федеральный университет, Владивосток, Россия;
4 Национальный научный центр морской биологии им. А.В. Жирмунского Дальневосточного отделения Российской академии наук, Владивосток, Россия
* Корреспондирующий автор (garbuzmihail.93[at]gmail.com)
АннотацияБолезнь Вильсона-Коновалова (БВК, WD) (OMIM 277900) или гепатолентикулярная дегенерация (ГЛД) представляет собой аутосомно-рецессивное заболевание, вызванное нарушением экскреции меди с ее последующим накоплением в печени, мозге и других тканях организма. В основе дефекта метаболизма меди лежит мутация в гене ATP7B, кодирующем транспортирующую медь АТФазу P-типа. Необычайный полиморфизм клинических проявлений и высокий процент бессимптомного течения патологии затрудняют своевременную постановку диагноза, задерживают начало курса терапии, обеспечивая неблагоприятный прогноз у больных и постановку диагноза у членов их семей. В таких условиях на ранних этапах развития болезни основным диагностическим критерием становится установление спектра мутаций гена ATP7B в каждом конкретном случае. Однако, разнообразие подходов к методам молекулярно-генетического исследования значительно затрудняет выбор единой адекватной методики. Целью настоящей работы является представление современных достижений в молекулярно-генетической диагностике ГЛД при разных мутациях в гене ATP7B для выбора наиболее перспективного метода диагностики при обследовании пациентов.
Ключевые слова: гепатолентикулярная дегенерация, молекулярно-генетическая диагностика.MOLECULAR GENETIC DIAGNOSIS OF HEPATOLENTICULAR DEGENERATION: FEATURES AND PROSPECTS
Review article
Garbuz M.M.1, *, Ovchinnikova E.V.2, Ovchinnikov A.V.3, Kumeyko V.V.4, Ovchinnikova A.A.5
1 ORCID: 0000-0003-0272-4478;
1-5 Far Eastern Federal University, Vladivostok, Russia;
4A.V. Zhirmunsky National Scientific Center of Marine Biology, Far Eastern branch of the Russian Academy of Sciences, Vladivostok, Russia
* Corresponding author (garbuzmihail.93[at]gmail.com)
AbstractWilson's disease (OMIM 277900), or hepatolenticular degeneration, is an autosomal recessive disease caused by impaired excretion of copper with its subsequent accumulation in the liver, brain, and other tissues of the body. The copper metabolism defect is based on a mutation in the ATP7B gene encoding the P-type copper-transporting ATPase. The extraordinary polymorphism of clinical manifestations and the high percentage of asymptomatic pathology complicate the timely diagnosis, delay the start of the course of therapy, contributing to an unfavorable prognosis in patients and diagnosis in their family members. In such conditions, at the early stages of the disease development, the main diagnostic criterion is to establish the spectrum of mutations of the ATP7B gene in each separate case. However, the variety of approaches to the methods of molecular genetic research makes choosing a single adequate methodology much more difficult. The purpose of this study is to present modern achievements in the molecular genetic diagnosis of hepatolenticular degeneration with different mutations in the ATP7B gene with the goal of selecting the most promising diagnostic method in the examination of patients.
Keywords: hepatolenticular degeneration, molecular genetic diagnostics.
Аббревиатуры
АТФ – Аденозинтрифосфат
АДФ – Аденозиндифосфат
АТФаза - Аденозинтрифосфатаза
ГЛД - гепатолентикулярная дегенерация
ПЦР – полимеразная цепная реакция
Несмотря на вековую давность истории изучения гепатолентикулярной дегенерации и разработку патогенетической терапии, до настоящего времени она остаётся заболеванием, которое деструктивно влияет на качество жизни пациентов, нередко приводит к утрате трудоспособности и летальному исходу.
Впервые патология была описана в 1883 году C. Westphal и A. Strumpell. Из-за сходства дрожательного гиперкинеза, возникающего у таких пациентов, с подобным дрожанием у больных рассеянным склерозом они именовали ее «псевдосклерозом». Свое современное название – гепатолентикулярная дегенерация (ГЛД) заболевание получило в честь Сэмюеля Вильсона, который в 1912 году, обнаружив у больных с дрожательным гиперкинезом дегенеративные изменения и в печени, и в лентикулярных ядрах головного мозга, предложил назвать заболевание - «прогрессирующей лентикулярной дегенерацией». В нашей стране учение о ГЛД связано с именем Н.В. Коновалова и представителей его школы, которые на основании многолетнего изучения данной патологии создали клиническую классификацию ее форм и оживили интерес к изучению ГЛД во всем мире [1].
В 1953 году A.G. Bearn установил аутосомно-рецессивный тип наследования болезни, в 1956 году - J.M. Walshe синтезировал из пенициллина препарат, оказывающий медьэлиминирующее действие – D-пеницилламин. В 1974 году - были приведены доказательства различных мутаций в гене ATP7B при развитии нарушений биллиарной экскреции меди у пациентов с ГЛД. В 1993 году ген ATP7B, ответственный за заболевание, удалось идентифицировать, локализовать на 13 хромосоме и сформулировать механизм развития патологии. Было установлено, что этот ген кодирует фермент - АТФ-азу Р типа, которая участвует как в выведении из организма с желчью излишка меди, так и во включении меди в апоцерулоплазмин для синтеза функционального церулоплазмина [2], [3]. Высвобождение свободной меди приводит к её отложению в различных органах и тканях. Заболевание проявляется в результате постепенного накопления меди в печени, мозге и тканях других органов, приводя к их повреждению и гибели клеток [4]. Различия в темпах накопления токсической меди в органах и индивидуальная чувствительность к их повреждению обеспечивают полиморфизм клинических проявлений болезни. Многочисленные клинические наблюдения указывали на зависимость клиники, от возраста дебюта, длительности течения и чувствительности тканей к токсичности меди. Были выделены формы болезни как с преимущественным поражением печени, почек, эндокринной системы, так и преобладанием неврологической симптоматики и психических нарушений. [5]. Поэтому сложность клинической диагностики длительно обеспечивала ее доступность только на этапе необратимых изменений в организме. Отсюда традиционно было принято считать ГЛД редким заболеванием, хотя уже в 2014 году P. Ferenci в своих исследованиях доказал, что до 40% детей и 58% взрослых на момент постановки диагноза уже имеют цирроз печени. [6]. Согласно официальной статистике выявляемость ГЛД составдяет 1:35 000–45 000 населения. Вместе с тем, периодически появляются указания на регионы с высокими показателями заболеваемости (превышающие традиционные в 3 - 4 раза) [7], [8]. В России ежегодная выявляемость ГЛД составляет 1:167000, при гетерозиготном носительстве 1:100 и расчётной распространённости - 1:10000, что указывает на проблемы диагностики данного заболевания [2].
Традиционно диагноз ГЛД устанавливается по результатам комплекса клинических, биохимических, гистологических и генетических показателей. При этом основной трудностью ранней диагностики ГЛД оказывается отсутствие единого критерия какого-либо из показателей, так как ни один из доступных биохимических тестов нельзя признать универсальным и специфичным для точного установления ГЛД. Кроме этого, стандартные лабораторные исследования могут давать ложноположительные или ложноотрицательные результаты. Задержка диагностики приводит к тому, что начало терапии запаздывает на годы. Поэтому обнаружение широкого спектра мутации гена АТР7В не только дало объяснение клиническому полиморфизму заболевания, но и вселило надежду на возможность его ранней диагностики с помощью совершенствования методик молекулярно-генетического обследования.
Роль АТФазы в метаболизме меди
Медь-транспортирующие АТФазы принадлежат к P1 (CPX-, P1B-) - подсемейству АТФаз P-типа. Члены этого подсемейства участвуют в транспорте различных переходных металлов (Cu, Ag, Cu2, Zn2, Ni2, Cd2, Pb2) через клеточные мембраны, в отличие от других АТФаз P-типа, которые переносят нетяжелые металлы (Na, K, Ca2, Mg2) или протонов [10]. В норме ATP7B выполняет две функции в печени. Во-первых, транспортирует медь в аппарат Гольджи для включения в церулоплазмин и экспортирует избыточную медь, связывая металл в везикулах для последующей экскреции с желчью. Вторая функция требует доставки ATP7B от аппарата Гольджи к эндоцитарным пузырькам в ответ на повышение внутриклеточной концентрации меди [2], [3].
В структуру ATP7B входят несколько регионов. «Хвост» на амино-конце содержит шесть металлсвязывающих доменов (ранее называемых медьсвязывающими единицами), каждый из которых имеет характерный аминокислотный мотив CXXC. Восемь трансмембранных сегментов образуют пору. Нуклеотид-связывающий участок находится в цитоплазме и состоит из трёх доменов: N-домен для связывания нуклеотидов АТФ / АДФ, P-домен (домен фосфатазы), где инвариантный остаток аспартата временно фосфорилируется, и адомен (активатор), облегчающий каталитический процесс. Карбокси-концевой участок частично неупорядочен и содержит мотив трилейцина, регулирующий внутриклеточный трафик [11] (см. рисунок 1).
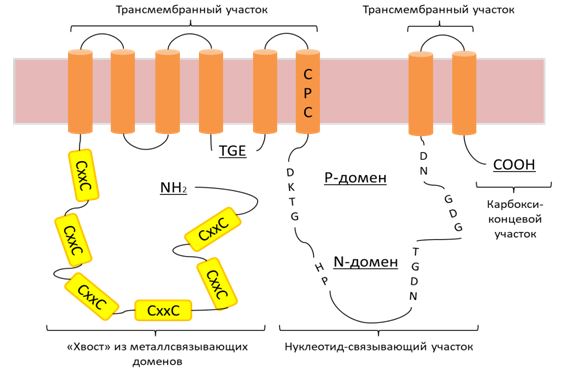
Рис. 1 – Строение и трансмембранная организация ATP7B
ATP7B - опосредованный транспорт меди включает несколько стадий. Во-первых, ATP7B связывает медь через свой цитозольный N-концевой металлсвязывающий домен и АТФ через нуклеотид-связывающий домен. Затем АТФ гидролизуется, и АТP7В временно фосфорилируется по остатку D1027, расположенному в Р-домене (каталитическое фосфорилирование). Последующее дефосфорилирование высвобождает энергию, необходимую для переноса меди через мембрану (этап переноса) (см. рисунок 2) [12].
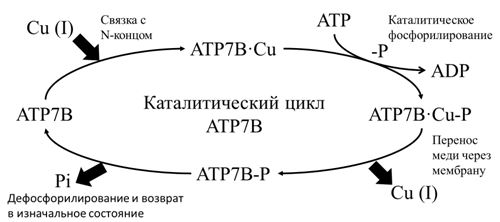
Рис. 2 – Схема каталитического цикла ATP7B
Зависимость метаболизма меди от мутации ATP7B
На каждый из перечисленных шагов транспорта меди могут влиять мутации ATP7B, вызывающие ГЛД [10]. Эффект может приводить к полной потере функции ATP7B, если мутированные остатки являются критическими для связывания АТФ или меди и / или конформационных переходов во время катализа. Инактивация ATP7B может быть частичной, если мутации снижают сродство к субстратам, замедляют конформационные переходы или мешают точному нацеливанию белка на аппарат Гольджи или везикулы. Понимание фенотипического разнообразия при ГЛД требует знания того, как мутации, вызывающие заболевание, изменяют стабильность, активность и локализацию белка в клетке. Однако, в настоящее время такая подробная информация недоступна для большинства мутаций, вызывающих ГЛД [12].
Мутации ATP7B можно разделить на следующие группы:
(1) миссенс-мутации, вызывающие одиночные аминокислотные замены в белковой последовательности,
(2) бессмысленные мутации, приводящие к вставке стоп-кодона и преждевременному прекращению трансляции,
(3) мутации со сдвигом рамки считывания, обычно вызванные делецией или вставкой нескольких нуклеотидов, и
(4) мутации сплайсинга и большие перестройки генов, которые приводят к грубой модификации или полной потере транскрипта.
Мутации 2–4 типов зачастую приводят к утрате способностей белка выполнять свои функции и гибели пациента на ранних этапах его жизни, поэтому внимание исследователей сконцентрировано на миссенс-мутациях [10]. Так же дефекты в гене ATP7B могут стать причиной накопления других металлов (железо, марганец) в нервной ткани пациентов с ГЛД [13].
Существует корреляция между мутациями в гене ATP7B, фенотипическими проявлениями и течением болезни. Некоторые мутации ATP7B препятствуют сворачиванию белка, вследствие чего он не попадает в цитоплазму из эндоплазматического ретикулума и деградирует. Самым распространённым примером таких мутаций является миссенс мутация H1069Q. Исследования invitro показали, что эта мутация делает невозможным свертывание NH-домена ATP7B при физиологической температуре, вызывая его деградацию в эндоплазматическом ретикулуме [14]. Это приводит к тому, что белок не способен достичь аппарата Гольджи и, следовательно, сайта экскреции меди. При данной мутации у пациентов наблюдаются низкие уровни церулоплазмина и высокие уровни свободной меди. У пациентов с заменой H1069Q было показано среднетяжелое течение заболевания с преимущественным проявлением психоневрологических симптомов в возрасте 20–22 лет [15], [16]. Оказалось, что кольца Кайзера-Флейшера встречаются у гомозиготных пациентов H1069Q чаще, чем у сложных гетерозиготных индивидуумов [17].
Вторым частым примером является мутация G875R, которая так же приводит к удержанию белка в эндоплазматическом ретикулуме. Задержка дефектного ATP7B в этих органеллах связана с тем, что для поступления белка в цитоплазму ему необходимо получить универсальный сигнал сортировки DKWSLLL, который присоединяется к C-концу белка. Однако было установлено, что добавление экзогенной меди в клеточную среду роста стабилизирует белок, позволяя ему завершить запланированную миграцию в аппарат Гольджи и преодолеть вызывающий болезнь фенотип. Теоретически, пациенты с этим конкретным вариантом могут быть более чувствительными к дефициту меди в рационе [18].
Соответственно мутации H1069Q и G875R препятствуют связыванию белка с DKWSLLL, что приводит к невозможности достижения ферментом ATP7B сайта экскреции меди, вызывая отложение меди в клетках и их токсическое поражение [19]. Однако исследования Lalioti с соавторами продемонстрировали частичное сохранение функции транспортировки меди, возможно, объясняя более мягкие фенотипы, связанные с определенными мутациями [20].
Среди мутаций, которые замедляют прохождение дефектного ATP7B через аппарат Гольджи, наиболее часто встречаемыми являются T977M в трансмембранном домене 6 и P1352S/L/R в 7 и 8 домене. Данные мутации соответствуют мутациям T994I и P1386S в ATP7A, которые вызывают тяжёлую болезнь Менкеса. Однако доказано, что они вызывают двигательные нарушения и снижение чувствительности. Это ещё раз демонстрирует всю сложность и многофакторность проявлений интоксикации медью [21], [22]. Мутация N41S приводит к нарушению сигнальной последовательности FAFDNVGY, которая отвечает за перенос базолатеральными эндосомами, в результате чего происходит накопление дефектного ATP7B в базолатеральной плазматической мембране клеток печени [23], [24].
Так же существует предположение о роли других генов, которые влияют на появление ГЛД и изменяют клинический фенотип. В число этих генов включены MTHFR [25], COMMD1 [26], ATOX1 [27], XIAP [28], PNPLA3 [29] и DMT1 [30]. Однако ни один из этих генов не продемонстрировал значительную диагностическую или прогностическую ценность. В 2013 году Coffey с соавторами провели исследование в Соединённом Королевстве и установили, что за проявление ГЛД отвечает только один ген ATP7B [8].
Так как, первопричинами появления ГЛД у человека является именно мутации гена ATP7B, кодирующего АТФазу Р-типа, то своевременному началу патогенетической терапии, должна способствовать быстрая и точная диагностика дефектов в этой первопричине.
Роль молекулярно-генетического тестирования в диагностике ГЛД
ГЛД принадлежит к группе заболеваний, которые требуют диагностики на ранних этапах развития болезни. Диагноз ГЛД определяется сочетанием клинических проявлений и лабораторных показателей, которые указывают на нарушение обмена меди с ее накоплением в печени и мозговой ткани – снижением содержания церулоплазмина в сыворотке крови и повышения суточной экскреции меди с мочой. Однако, эти стандартные тесты на раннем этапе заболевания могут давать как ложноположительные, так и ложноотрицательные результаты. Невозможность диагностировать пациента с ГЛД на раннем этапе заболевания может привести или к потере возможностей патогенетической терапии, или к несоответствующему введению потенциально токсичных препаратов таким пациентам при ложноположительной диагностике [5], [6].
Клиническая диагностика ГЛД затруднена в связи с широким спектром клинических проявлений и с отсутствием типичных симптомов у пациентов с этим заболеванием. Подозрение на ГЛД должно возникать в каждом случае необъяснимой печёночной, неврологической или психической дисфункции. При этом, методы и тесты для диагностики ГЛД значительно отличаются у пациентов с печёночной дисфункцией и психоневрологической симптоматикой [5], [6].
В настоящее время ни один из доступных лабораторных тестов нельзя признать универсальным и специфичным для ранней диагностики ГЛД [5]. Для точной постановки диагноза используется соответствие критериям Лейпцига, которые были приняты на 8-м Международном совещании по ГЛД и болезни Менкеса и приняты в Европейской ассоциации по изучению печени [5]. Ключевыми клиническими диагностическими признаками, которые легли в основу формирования критериев Лейпцига, стали признаки печеночной дисфункции, острый гемолиз с почечной недостаточностью, расстройства двигательных функций, нервно-психические расстройства и кольца Кайзера-Флейшера на роговице [31]. Данная система оценки ГЛД обеспечивает хорошую диагностическую точность у пациентов с ярко выраженными симптомами болезни, но не способна продемонстрировать высокую эффективность у пациентов на ранних этапах заболевания или у их родственников [32]. Поэтому внедрение методов молекулярно-генетической диагностики, которые позволяют выявить первопричину на любых этапах течения болезни дало надежду на ранее решение диагностических вопросов у пациентов с ГЛД.
Методы молекулярно-генетической диагностики ГЛД
Среди наиболее распространенных методов молекулярно-генетического исследования, используемых для диагностики ГЛД, наиболее распространенными являются:
- Секвенирование гена ATP7B по Сэнгеру;
- Генетическая диагностика ГЛД, основанная на амплификации ДНК методом ПЦР;
- RestrictionFragmentLengthPolymorphism или Полиморфизм длин рестрикционных фрагментов (ПДРФ-анализ);
- Single-strandconformationpolymorphismanalysis или анализ одноцепочечного конформационного полиморфизма (SSCP-анализ);
- Multiplexligationprobeamplification или мультиплексная амплификация лигированных зондов (MLPA-анализ);
- ARMSAmplificationRefractoryMutationSystem или аллель-специфическая ПЦР в режиме реального времени (ARMS);
- HighResolutionMelting или Анализ Кривых Плавления с Высоким Разрешением (HRM);
- TaqManAssay или Выщепление 5' концевой метки (система TaqMan);
- Использование микрочипа;
- Методы молекулярно-генетической диагностики ГЛД основанные на next-generationsequencing.
Далее рассмотрим каждый из методов подробнее.
Секвенирование гена ATP7B по Сэнгеру
Прямая молекулярно-генетическая диагностика затруднена из-за наличия 800 возможных мутаций в гене ATP7B (http://www.wilsondisease.med.ualberta.ca/). Однако тип мутации играет роль в прогнозировании клинических проявлений. Так же, несколько частых мутаций демонстрируют своеобразное глобальное распределение [33]. Кроме того, большинство пациентов являются сложными гетерозиготами (то есть они несут две разные мутации).Секвенирование по Сэнгеру гена ATP7B, позволяет выявлять установленные мутации и у членов семьи больного. Благодаря высокой надежности и высокоинформативным результатам этот метод заслужил широкое распространение в большинстве стран мира. В настоящее время секвенирование ДНК по Сэнгеру полностью автоматизировано и проводится на специальных приборах, секвенаторах [34].Для проведения секвенирования необходимо пройти множество этапов: выделить ДНК из целевого органа или его части, провести ПЦР, очистить смесь для дальнейшего использования, провести пробоподготовку для секвенирования и само секвенирование. Такая многоэтапность увеличивает вероятность ошибки, является достаточно дорогой из-за использования большого количества лабораторного оборудования и увеличивает время исследования. Поэтому в настоящее время секвенирование по Сенгеру не используется широко в целях диагностики заболевания, но благодаря, высокой точности и информативности он остается «золотым стандартом» и применяется для подтверждения результатов других методов диагностики [35], [36], [37].
В медицинской практике, помимо секвенирования по Сенгеру, в последние десятилетия были предложены методы молекулярно-генетической диагностики ГЛД основанные на амплификации ДНК методом ПЦР. ПЦР (или полимеразная цепная реакция) – метод молекулярной биологии, позволяющий добиться увеличения малых концентраций целевого фрагмента нуклеиновой кислоты (ДНК или РНК) в пробе. ПЦР менее требовательна к реактивам и умениям в сравнении с большинством других молекулярных методов [38], амплификаторы доступны для большинства обычных лабораторий. А высокая специфичность праймеров была достигнута с помощью 3'-нуклеотида, сопоставленного с последовательностями, специфичными для мутации. Возможность обнаружения генотипоспецифических мутаций в сочетании с экономичным использованием красителей для детекции ДНК и мониторингом пороговых значений флюоресценции эти праймеры позволяли специфически амплифицировать последовательности-мишени [39], [40]. Ранее наличие неспецифических продуктов ПЦР являлось ограничением и должно было тщательно проверяться с помощью гель-электрофореза, что увеличивало время получения результатов. Однако ПЦР с горячим стартом и буферы, содержащие сбалансированную комбинацию катионов для стимулирования специфического отжига, устранили данную проблему. Время, сэкономленное с помощью методов диагностики, основанных на ПЦР, позволяло быстро подтвердить наличие заболевания у пациентов с угрозой молниеносной печеночной недостаточности или с бессимптомным течением [39].
Одним из первых методов, разработанных для анализа на амплификаторе, является ПДРФ-анализ (RestrictionFragmentLengthPolymorphism или Полиморфизм длин рестрикционных фрагментов). Это метод, который использует вариации в гомологичных последовательностях ДНК. ПДРФ-анализ заключается в дроблении образца на части в специфических местах с использованием фрагментов рестрикции и детектированием их длин с помощью гель-электрофореза и переносом на мембрану с помощью саузерн-блота. Во время саузерн-блота происходит гибридизация мембраны с меченным ДНК-зондом, с помощью которого сравниваются длины полученного фрагмента и контроля. Несмотря на то, что данный метод в значительной степени устарел, он до сих пор применяется за счёт своей невысокой цены и простоты[41], [42].
SSCP-анализ (single-strandconformationpolymorphismanalysis или анализ одноцепочечного конформационного полиморфизма) – быстрый метод оценки последовательностей ДНК основанный на ПЦР. Данный метод заключается в амплификации изучаемого гена длиной несколько сотен (200-300 п.н.) нуклеотидов, его последующей денатурации и электрофорезе. В процессе анализа электрофореграмм мутации выявляются за счёт конформационных изменений ДНК. Сам принцип метода состоит в том, что фрагменты ДНК с разной конформацией имеют разную скорость при прохождении электрофорезного геля. Это проявляется в сдвиге полосы амплифицированного фрагмента мутантной ДНК по отношению к ДНК дикого типа. SSCP-анализ актуален для выявления замен оснований, малых инсерций и делеций, различных перестроек, а также для поиска новых мутаций. Плюсами SSCP-анализа является возможность изучения большого количества проб одновременно, что позволяет анализировать биологический материал большого числа больных. Высокая чувствительность метода позволяет выявлять до 90-95% всех мутаций. Однако малый размер анализируемых фрагментов усложняет поиск нескольких мутаций, особенно при большом размере целевого гена. Для решения данной проблемы приходится увеличивать количество образцов, что приводит к увеличению стоимости и времени исследования [43].
MLPA-анализ (Multiplexligationprobeamplification или мультиплексная амплификация лигированных зондов) – это разновидность мультиплексной ПЦР, которая позволяет амплифицировать несколько (до 60 зондов) образцов только с одной парой праймеров. После денатурации ДНК образца к нему добавляют смесь MLPA-зондов, которые состоят из двух олигонуклеотидов. Если в образце присутствует последовательность комплементарная зонду, то олигонуклеотиды гибридизируются друг за другом так, чтобы быть сшитыми лигазой в единый зонд после чего зонды разной длины амплифицируются. В результате происходит амплификация не ДНК образца, а смеси зондов уникальной длины (от 130 до 500 п.о.). Так же один из праймеров несёт флуорисцентную метку для визуализации продуктов амплификации в ходе капиллярного электрофореза. При наличии мутаций в исследуемом образце высота его пиков при анализе будет ниже, чем высота пиков референсного образца. MLPA является высокочувствительным методом и позволяет одновременно определять относительно большое количество копий всех экзонов. Однако, в конечном этапе результат часто зависит от опыта лабораторного работника и является субъективным. При использовании MLPA сложнее обнаружить однонуклеотидные замены [44].
Одним из тестов, использующих амплификатор и часто применяемый для диагностики ГЛД является ARMS (AmplificationRefractoryMutationSystem или аллель-специфическая ПЦР в режиме реального времени). Типичный тест ARMS состоит из двух дополнительных реакций. Первая реакция содержит праймер ARMS, который специфичен для нормальной последовательности ДНК и не может амплифицировать мутантную ДНК в определенном локусе. Точно так же вторая реакция содержит специфический мутантный праймер и не амплифицирует нормальную ДНК. Таким образом, нормальные индивидуумы генерируют продукт ПЦР только в нормальной реакции, гетерозиготы генерируют продукты в обеих реакциях, а гомозиготные индивидуумы-мутанты делают это только в мутантной реакции. Однако, данный метод не подходит для диагностики пациентов с ГЛД в регионах с плохо изученным спектром мутаций из-заневозможности обнаружения новых мутаций [41], [45], [46].
Технология метода HRM (HighResolutionMelting или Анализ Кривых Плавления с Высоким Разрешением)базируется на гетеродуплексном анализе с использованием интеркалирующего красителя и состоит из двух этапов. Первый этап представлен ПЦР, которая служит для накопления ампликонов, содержащих образцы дикого типа и мутантные последовательности нуклеотидов (SNP). Второй этап заключается в снижении температуры до 25°С для формирования низкоспецифичных дуплексов. В результате формируется два вида дуплексов: высокоспецифичные или полностью комплементарные гомодуплексы (гомогенные дуплексы, например «нормальный» ампликон + «нормальный» ампликон; мутантный ампликон + мутантный ампликон); низкоспецифичные гетеродуплексы (гетерогенные: «нормальный» ампликон + мутантный ампликон). В этот же момент происходит встраивание интеркалирующего красителя между цепями дуплекса и повышение флуоресценции. Далее температуру начинают постепенно повышать, в результате чего плавятся цепи дуплекса и высвобождается интеркалирующий краситель. На данном этапе флюоресценция убывает и происходит её фиксирование. Для проведения анализа используется температура плавления каждого вида дуплексов, что, в сочетании с уровнем флуоресценции, позволяет их идентифицировать. С помощью HRM-анализа удается не только идентифицировать уже известные мутации, но и обнаруживать новые однонуклеотидные замены [38]. Продукты ПЦР-амплификации, полученные из анализа HRM, могут быть непосредственно использованы для прямого секвенирования без какой-либо предварительной обработки. Однако малый шаг увеличения температуры на втором этапе исследования приводил к необходимости математической обработки результатов, увеличивая возможность ошибки в их интерпретации из-за минимальных различий кривых плавления для разных генотипов [41].
Ещё одним часто используемым методом диагностики является система TaqMan (TaqManAssay или Выщепление 5' концевой метки). Она отличается относительной простотой, высокой специфичностью и представляет собой линейный олигонуклеотид, меченый по 5'-концу флуорофором, а по 3'-концу – гасителем флуоресценции. Принцип зонда TaqMan основан на 5'-3'-экзонуклеазной активности Taq-полимеразы для расщепления двойного меченого зонда во время гибридизации с комплементарной последовательностью-мишенью и обнаружения мутаций на основе флуорофора. Как и в других количественных методах ПЦР, полученный сигнал флуоресценции позволяет количественно измерять накопление продукта во время экспоненциальных стадий ПЦР, зонд TaqMan значительно повышает специфичность обнаружения [47]. Однако необходимость конструирования зондов и оптимизации условий ПЦР является недостатком этого метода [41].
Несмотря на высокую точность генотипирования, воспроизводимость и потенциал для быстрого и высокопроизводительного анализа в клинических лабораториях, при проведении диагностики для всех вышеперечисленных методов необходим контрольный образец дикого типа, который служит для поиска мутаций и обнаружения неспецифической амплификации. В качестве контрольного образца дикого типа используют геномную ДНК от 100 здоровых людей данной популяции без ГЛД. Такое количество необходимо чтобы исключить возможность попадания вновь выявленных мутаций в выборку. Данное условие сильно усложняет проведение диагностики, особенно в регионах со слабоизученным распределением мутаций [48], [49].
Простым и быстрым способом провести скрининг мутаций ATP7B является микрочип с генотипированием. Данный чип способен одновременно обнаруживать множество мутаций и полиморфизмов в гене ATP7B на основе реакции удлинения массива праймеров. ДНК пациента амплифицируется в результате четырех мультиплексных полимеразных цепных реакций, фрагментированные продукты отжигаются с матричными праймерами, нанесенными на чип, что позволяет проводить реакции удлинения ДНК-полимеразы с флуоресцентно меченными дидезоксинуклеотидами. Микрочип Уилсона был проверен путем скрининга 97 ранее генетически подтвержденных пациентов с ГЛД и продемонстрировал 100% точностью при скрининге. Данный чип является быстрым, чувствительным и экономически эффективным инструментом, представляющим собой прототип чипа, который облегчает и ускоряет скрининг потенциальных пациентов с ГЛД. В данном чипе так же встроен положительный контроль, что избавляет данный метод от необходимости подбора и создания контрольной группы. Однако, данная технология позволяет обнаруживать только выбранный спектр известных мутаций, результаты часто зависят от условий эксперимента и оборудования, поэтому требуется очень точная оптимизация, что увеличивает требования к навыкам специалиста, который будет проводить скрининг [48].
Методы молекулярно-генетической диагностики ГЛД основанные на next-generationsequencing.Ранние исследования отдельных экзонов с использованием секвенирования по Сенгеру не смогли идентифицировать мутации в гене ATP7B в значительном числе клинически диагностированных случаев. Это привело к обеспокоенности по поводу генетической гетерогенности ГЛД и предполагало наличие необычных мутационных механизмов. Однако в крупном исследовании с использованием next-generationsequencing (NGS) Coffeyetal. представили результаты исследования у 181 пациента с клинически и биохимически подтверждённым диагнозом ГЛД. Всего в ходе анализа было обнаружено 116 различных мутаций ATP7B, 32 из которых являются новыми. Общая точность обнаружения мутаций составила 98%. В ходе исследования было выявлено, что вероятность мутаций в генах, отличных от ATP7B, вызывающих фенотип ГЛД, очень мала, что подтверждает моногенный тип наследования ГЛД. Данный метод имеет значение для анализа и исследования гена ATP7B. В клинической практике это даёт возможность учитывать необычные генетические механизмы, такие как диспаратная дисомиология или возможное присутствие трех мутаций ATP7B, что может способствовать прогнозированию течения заболевания и возможности лучшего подбора терапии. Однако, несмотря на все преимущества NGS метода, он является самым дорогостоящим среди всех используемых методов молекулярной диагностики ГЛД [8].
Заключение
Разнообразие методов молекулярно-генетической диагностики при использовании оборудования разной сложности и стоимости свидетельствует не столько об интересе исследователей к молекулярно-генетической диагностике ГЛД, сколько об отсутствии единого универсального подхода к их выбору в разных странах мира. Отчетливо прослеживается зависимость использования того или иного метода от клинических проявлений различных форм патологии, стратегии предполагаемого лечения и стоимости оборудования. Например, NGS, несмотря на высокую стоимость, более целесообразен для исследования механизмов экспрессии и строения генов ATP7B в небольших, но высоко развитых странах, где есть возможность организовывать централизованный сбор образцов генетического материала за короткие сроки, что способно обеспечить быстрое подтверждение диагноза. В странах с большими территориями преимуществом обладают методы, использующие амплификаторы и микрочипы. Данная категория методов молекулярной диагностики является самой дешевой из вышеперечисленных вариантов, а также не требует от исследователя особых навыков для проведения процедуры. Создание нескольких центров для сбора и анализа материала на больших территориях позволит оперативно и эффективно определять наличие мутаций у пациентов с подозрением на ГЛД. Наиболее усреднённым методом по стоимости, требованию к навыкам, скорости и эффективности проведения принято считать секвенирование по Сенгеру. Этот метод доступен к применению в любых странах, в частности и в тех, в которых ещё не установлен спектр мутаций гена ATP7B. Поэтому в настоящее время золотым стандартом молекулярно-генетической диагностики ГЛД признан именно этот метод.
Таким образом, наиболее перспективным и универсальным инструментом для диагностики ГЛД следует признать молекулярно-генетическое тестирование. Это обусловлено тем, что начало диагностического процесса с молекулярного тестирования может значительно снизить потребность в инвазивных методах обследования, способствовать точной постановке диагноза у пациентов на ранних этапах заболевания или избежать ложноположительных результатов, приводящих к назначению неадекватного лечения с развитием гематологических и неврологические осложнений [50].
| Конфликт интересов Не указан. | Conflict of Interest None declared. |
Список литературы / References
- Еремина Е. Ю. Болезнь Вильсона-Коновалова / Е. Ю. Еремина // Вестник современной клинической медицины. – 2011. - Т. 1. - №4 – С. 38-46.
- Bull P.C. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene / P.C. Bull et al. // Nat Genet. – 1993. – V.4. - №5 – P.327-337. DOI: 10.1038/ng1293-327.
- Compston A. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver, by S. A. Kinnier Wilson, (From the National Hospital, and the Laboratory of the National Hospital, Queen Square, London) / A. Compston // Brain. – 2009. – V.8. - №132 – P.1997-2001. DOI: 10.1093/brain/awp193.
- Lalioti V. Disorders in Hepatic Copper Secretion: Wilson's Disease and Pleomorphic Syndromes / V. Lalioti et al. // Semin Liver Dis. – 2017. – V.2. - №37 – P.175-188. DOI: 10.1055/s-0037-1602764.
- European Association for Study of Liver. EASL clinical practice guidelines: Wilson’s disease / EASL //J Hepatol – 2012. – V.3. - №56 – P.671–
- Ferenci P. Age and Sex but Not ATP7B Genotype Effectively Influence the Clinical Phenotype of Wilson Disease / Ferenci et al. // Hepatology – 2019. – V.4. - №69 – P.1464-1476. DOI: 10.1002/hep.30280.
- Shimizu N. Effects of long-term zinc treatment in Japanese patients with Wilson disease: efficacy, stability, and copper metabolism / N. Shimizu et al. // Transl Res. – 2010. – V.6. - №156 - P.350-357. DOI: 10.1016/j.trsl.2010.08.007.
- Coffey A.J. A genetic study of Wilson's disease in the United Kingdom / A.J. Coffeyet al. // Brain. – 2013. – V.5.- №136 – P.1476-1487. DOI: 10.1093/brain/awt035.
- Баязутдинова Г.М. Мутация с.3207C>A гена АТР7В – наиболее частая причина гепатолентикулярной дегенерации в России: частота и причина распространения / Г.М. Баязутдинова, О.А.Щагина, А.В.Поляков // Медицинская генетика. – 2018. - Т. 4. - №17 – С. 25-30.
- Tsivkovskii R. Copper-Transporting ATPases: Key Regulators of Intracellular Copper Concentration / Tsivkovskii et al. // Handbook of ATPases – 2005. – P.99–158.DOI:10.1002/3527606122.ch5
- Jayakanthan S. Human copper transporter ATP7B (Wilson disease protein) forms stable dimers in vitroand in cells / Jayakanthan et al. // J Biol Chem. – 2017. - V.46. - №17 - P.18760-18774. DOI: 10.1074/jbc.M117.807263.
- Huster D. Diverse functional properties of Wilson disease ATP7B variants / D. Huster et al. // Gastroenterology. – 2012. – V.4. - №142 - P:947-956. DOI: 10.1053/j.gastro.2011.12.048.
- Dusek P. The neurotoxicity of iron, copper and manganese in Parkinson's and Wilson's diseases / P. Dusek et al. // J Trace Elem Med Biol. – 2015. - №31 – P. 193-203. DOI: 10.1016/j.jtemb.2014.05.007.
- Dmitriev O.Y. Difference in stability of the N-domain underlies distinct intracellular properties of the E1064A and H1069Q mutants of copper-transporting ATPase ATP7B / O.Y. Dmitriev et al. // J Biol Chem. – 2011. – V.18. - №286 – 16355-16362. DOI: 10.1074/jbc.M110.198101.
- Stapelbroek J.M. The H1069Q mutation in ATP7B is associated with late and neurologic presentation in Wilson disease: results of a meta-analysis / J.M. Stapelbroek et al. // J Hepatol. – 2004. – V.5. - №41 – P.758-763. DOI: 10.1016/j.jhep.2004.07.017.
- Kalita J. R778L, H1069Q, and I1102T mutation study in neurologic Wilson disease / J. Kalita et al. // Neurol India. – 2010. – V.4. - №58 – P.627-630. DOI: 10.4103/0028-3886.68678.
- Folhoffer A. Novel mutations of the ATP7B gene among 109 Hungarian patients with Wilson's disease / A. Folhoffer et al. // Eur J Gastroenterol Hepatol. – 2007. – V.2. - №19 – P.105-111. DOI: 10.1097/01.meg.0000223904.70492.0b.
- Gupta A.Cellular copper levels determine the phenotype of the Arg875 variant of ATP7B/Wilson disease protein / Gupta et al. // Proc Natl Acad Sci USA. – 2011. – V.13. - №108 - P.5390-5395. DOI: 10.1073/pnas.1014959108.
- Braiterman L. Critical roles for the COOH terminus of the Cu-ATPase ATP7B in protein stability, trans-Golgi network retention, copper sensing, and retrograde trafficking / L. Braiterman et al. // Am J Physiol Gastrointest Liver Physiol. – 2011. – V.1. - №301 - P.69-81. DOI: 10.1152/ajpgi.00038.2011.
- Lalioti V. DKWSLLL, a versatile DXXXLL-type signal with distinct roles in the Cu(+)-regulated trafficking of ATP7B / V. Lalioti et al. // Traffic. – 2014. – V.8. - №15 – P.839-860. DOI: 10.1111/tra.12176.
- Dale J.M. X-linked spinal muscular atrophy in mice caused by autonomous loss of ATP7A in the motor neuron / L. Hodgkinson et al. // J Pathol. – 2015. – V.2. - №236 – P.241-250. DOI: 10.1002/path.4511.
- Kennerson M.L. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy / M.L. Kennerson et al. // Am J Hum Genet. – 2010. – V.3. - №86 – P.343-352. DOI: 10.1016/j.ajhg.2010.01.027.
- Braiterman L. Apical targeting and Golgi retention signals reside within a 9-amino acid sequence in the copper-ATPase, ATP7B / L. Braiterman et al. // Am J Physiol Gastrointest Liver Physiol. – 2009. – V.2. - №296 – P.433-444. DOI: 10.1152/ajpgi.90489.2008.
- Donoso M. Polarized traffic of LRP1 involves AP1B and SNX17 operating on Y-dependent sorting motifs in different pathways / M. Donoso et al. // Mol Biol Cell. – 2009. – V.1. - №20 – P.481-497. DOI: 10.1091/mbc.e08-08-0805.
- Gromadzka G. Genetic variability in the methylenetetrahydrofolate reductase gene (MTHFR) affects clinical expression of Wilson's disease / G. Gromadzka et al. // J Hepatol. – 2011. – V.4. - №55 – P.913-919. DOI: 10.1016/j.jhep.2011.01.030.
- Weiss K.H. Copper-induced translocation of the Wilson disease protein ATP7B independent of Murr1/COMMD1 and Rab7 / K.H. Weiss et al. // Am J Pathol. – 2008. – V.6. - №173 – P.1783-1794. DOI: 10.2353/ajpath.2008.071134.
- Simon I. Analysis of the human Atox 1 homologue in Wilson patients / I. Simon et al. // World J Gastroenterol. – 2008. – V.15. - №14 – P.2383-2387. DOI: 10.3748/wjg.14.2383.
- Weiss K.H. Genetic analysis of BIRC4/XIAP as a putative modifier gene of Wilson disease / K.H. Weiss et al. // J Inherit Metab Dis. – 2010. - №33 – P.233-240. DOI: 10.1007/s10545-010-9123-5.
- Stättermayer A.F. Hepatic steatosis in Wilson disease--Role of copper and PNPLA3 mutations / A.F. Stättermayer et al. // J Hepatol. – 2015. – V.1. - №63 – P.156-163. DOI: 10.1016/j.jhep.2015.01.034.
- Przybyłkowski A. Polymorphisms of metal transporter genes DMT1 and ATP7A in Wilson's disease / A. Przybyłkowski et al. // J Trace Elem Med Biol. – 2014. – V.1. - №28 – P.8-12. DOI: 10.1016/j.jtemb.2013.08.002.
- Ferenci P. Diagnosis and phenotypic classification of Wilson disease / P. Ferenci et al. // Liver Int. – 2003. –V.3. - №23 – P.139-142. DOI: 10.1034/j.1600-0676.2003.00824.x.
- Nicastro E. Re-evaluation of the diagnostic criteria for Wilson disease in children with mild liver disease / E. Nicastro et al. // Hepatology. – 2010. – V.6. - №52 – P.1948-1956. DOI: 10.1002/hep.23910.
- Ferenci P. Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing / P. Ferenci // Hum Genet. – 2006. – V.2. - №120 – P.151-159. DOI: 10.1007/s00439-006-0202-5.
- Sanger F. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase / F. Sanger et al. // Journal of molecular biology. – 1975. – V.3. - №94 – P.441-448.
- Chang I.J. The genetics of Wilson disease / I.J. Chang et al. // Handb Clin Neurol. – 2017 - №142 – P.19-34. DOI: 10.1016/B978-0-444-63625-6.00003-3.
- Bost M. Molecular analysis of Wilson patients: direct sequencing and MLPA analysis in the ATP7B gene and Atox1 and COMMD1 gene analysis / M. Bost et al. // J Trace Elem Med Biol. – 2012. - №26 – P.97-101. DOI: 10.1016/j.jtemb.2012.04.024.
- Lepori M.B. Mutation analysis of the ATP7B gene in a new group of Wilson's disease patients: contribution to diagnosis / M.B. Lepori et al. // Mol Cell Probes. – 2012. – V.4. - №26 – P.147-150. DOI: 10.1016/j.mcp.2012.03.007.
- Lin C.W. Development of a high-resolution melting method for the screening of Wilson disease-related ATP7B gene mutations / C.W. Lin et al. // Clin Chim Acta. – 2010. – V.17-18. - №411 – P.1223-1231. DOI: 10.1016/j.cca.2010.04.030.
- Mak C.M. Diagnosis of Wilson's disease: a comprehensive review. / C.M. Mak et al. // Crit Rev Clin Lab Sci. – 2008 – V.3. - №45 – P.263-290. DOI: 10.1080/10408360801991055.
- Gomes A. Geographic distribution of ATP7B mutations in Wilson disease / A. Gomes et al. // Ann Hum Biol. – 2016. – V.1. - №43 – P.1-8. DOI: 10.3109/03014460.2015.1051492.
- Mahdieh N. An overview of mutation detection methods in genetic disorders / N. Mahdieh et al. // Iran J Pediatr. – 2013. – V.4. - №23 – P.375-388.
- Arnheim N. Use of pooled DNA samples to detect linkage disequilibrium of polymorphic restriction fragments and human disease: studies of the HLA class II loci / N. Arnheim et al. // Proc Natl Acad Sci USA. – 1985. –V.20. - №82 – 6970-6974. DOI: 10.1073/pnas.82.20.6970.
- Hayashi K. How sensitive is PCR-SSCP / K. Hayashi et al. // Hum Mutat. – 1993. – V.5. - №2 – P.338-346. DOI: 10.1002/humu.1380020503.
- Schouten J.P. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification / J.P. Schouten et al. // Nucleic Acids Res. – 2002. – V.12. - №30 – P.57. DOI: 10.1093/nar/gnf056.
- Newton C.R. Amplification refractory mutation system for prenatal diagnosis and carrier assessment in cystic fibrosis / C.R. Newton et al. // Lancet. – 1989. – V.2. - №30 – P.1481-1483. DOI: 10.1016/s0140-6736(89)92931-0.
- Bathelier C. ARMS test for diagnosis of factor V Leiden mutation and allele frequencies in France / C. Bathelier et al. // Mol Cell Probes. – 1998. – V.2. - №12 – P.121-123. DOI: 10.1006/mcpr.1997.0152.
- Zappu A. Development of TaqMan allelic specific discrimination assay for detection of the most common Sardinian Wilson's disease mutations. Implications for genetic screening / A. Zappu et al. // Mol Cell Probes. –2010. – V.4. - №24 – 233-235. DOI: 10.1016/j.mcp.2010.01.004.
- Gojová L. Genotyping microarray as a novel approach for the detection of ATP7B gene mutations in patients with Wilson disease / L. Gojová et al. // Clin Genet. – 2008. – V.5. - №73 – P.441-452. DOI: 10.1111/j.1399-0004.2008.00989.x.
- Lu Y. Clinical and molecular characterization of Wilson's disease in China: identification of 14 novel mutations / H. Li et al. // BMC Med Genet. – 2011. – №12 - P.6. DOI: 10.1186/1471-2350-12-6.
- Kumar N. Myelopathy due to copper deficiency following gastrointestinal surgery / N. Kumar et al. // Arch Neurol. – 2003. – V.12. - №60 – P.1782-1785. DOI: 10.1001/archneur.60.12.1782. PMID: 14676057.
Список литературы на английском языке / References in English
- Yu. Bolezn' Vil'sona-Konovalova [Wilson-Konovalov disease] / E. Yu. Eremina // Vestnik sovremennoj klinicheskoj mediciny[Vestnik sovremennoy klinicheskoy meditsiny]. – 2011. - V. 1. - №4 – P. 38-46. [in Russian]
- Bull P.C. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene / P.C. Bull et al. // Nat Genet. – 1993. – V.4. - №5 – P.327-337. DOI: 10.1038/ng1293-327.
- Compston A. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver, by S. A. Kinnier Wilson, (From the National Hospital, and the Laboratory of the National Hospital, Queen Square, London) / A. Compston // Brain. – 2009. – V.8. - №132 – P.1997-2001. DOI: 10.1093/brain/awp193.
- Lalioti V. Disorders in Hepatic Copper Secretion: Wilson's Disease and Pleomorphic Syndromes / V. Lalioti et al. // Semin Liver Dis. – 2017. – V.2. - №37 – P.175-188. DOI: 10.1055/s-0037-1602764.
- European Association for Study of Liver. EASL clinical practice guidelines: Wilson’s disease / EASL // J Hepatol – 2012. – V.3. - №56 – P.671–
- Ferenci P. Age and Sex but Not ATP7B Genotype Effectively Influence the Clinical Phenotype of Wilson Disease / Ferenci et al. // Hepatology – 2019. – V.4. - №69 – P.1464-1476. DOI: 10.1002/hep.30280.
- Shimizu N. Effects of long-term zinc treatment in Japanese patients with Wilson disease: efficacy, stability, and copper metabolism / N. Shimizu et al. // Transl Res. – 2010. – V.6. - №156 - P.350-357. DOI: 10.1016/j.trsl.2010.08.007.
- Coffey A.J. A genetic study of Wilson's disease in the United Kingdom / A.J. Coffeyet al. // Brain. – 2013. – V.5.- №136 – P.1476-1487. DOI: 10.1093/brain/awt035.
- Bayazutdinova G.M. Mutacija s.3207C>A gena ATR7V – naibolee chastaja prichina gepatolentikuljarnoj degeneracii v Rossii: chastota i prichina rasprostranenija [The mutation p. 3207C> A of the ATР7B gene is the most common cause of hepatolenticular degeneration in Russia: frequency and cause of spread] / G.M. Bayazutdinova et al. // Meditsinskaya genetika. – 2018. - 4. - №17 – P. 25-30. DOI: 10.25557/2073-7998.2018.04.25-30. [in Russian]
- Tsivkovskii R. Copper-Transporting ATPases: Key Regulators of Intracellular Copper Concentration / Tsivkovskii et al. // Handbook of ATPases – 2005. – P.99–158.DOI:10.1002/3527606122.ch5
- Jayakanthan S. Human copper transporter ATP7B (Wilson disease protein) forms stable dimers in vitroand in cells / Jayakanthan et al. // J Biol Chem. – 2017. - V.46. - №17 - P.18760-18774. DOI: 10.1074/jbc.M117.807263.
- Huster D. Diverse functional properties of Wilson disease ATP7B variants / D. Huster et al. // Gastroenterology. – 2012. – V.4. - №142 - P:947-956. DOI: 10.1053/j.gastro.2011.12.048.
- Dusek P. The neurotoxicity of iron, copper and manganese in Parkinson's and Wilson's diseases / P. Dusek et al. // J Trace Elem Med Biol. – 2015. - №31 – P. 193-203. DOI: 10.1016/j.jtemb.2014.05.007.
- Dmitriev O.Y. Difference in stability of the N-domain underlies distinct intracellular properties of the E1064A and H1069Q mutants of copper-transporting ATPase ATP7B / O.Y. Dmitriev et al. // J Biol Chem. – 2011. – V.18. - №286 – 16355-16362. DOI: 10.1074/jbc.M110.198101.
- Stapelbroek J.M. The H1069Q mutation in ATP7B is associated with late and neurologic presentation in Wilson disease: results of a meta-analysis / J.M. Stapelbroek et al. // J Hepatol. – 2004. – V.5. - №41 – P.758-763. DOI: 10.1016/j.jhep.2004.07.017.
- Kalita J. R778L, H1069Q, and I1102T mutation study in neurologic Wilson disease / J. Kalita et al. // Neurol India. – 2010. – V.4. - №58 – P.627-630. DOI: 10.4103/0028-3886.68678.
- Folhoffer A. Novel mutations of the ATP7B gene among 109 Hungarian patients with Wilson's disease / A. Folhoffer et al. // Eur J Gastroenterol Hepatol. – 2007. – V.2. - №19 – P.105-111. DOI: 10.1097/01.meg.0000223904.70492.0b.
- Gupta A.Cellular copper levels determine the phenotype of the Arg875 variant of ATP7B/Wilson disease protein / Gupta et al. // Proc Natl Acad Sci USA. – 2011. – V.13. - №108 - P.5390-5395. DOI: 10.1073/pnas.1014959108.
- Braiterman L. Critical roles for the COOH terminus of the Cu-ATPase ATP7B in protein stability, trans-Golgi network retention, copper sensing, and retrograde trafficking / L. Braiterman et al. // Am J Physiol Gastrointest Liver Physiol. – 2011. – V.1. - №301 - P.69-81. DOI: 10.1152/ajpgi.00038.2011.
- Lalioti V. DKWSLLL, a versatile DXXXLL-type signal with distinct roles in the Cu(+)-regulated trafficking of ATP7B / V. Lalioti et al. // Traffic. – 2014. – V.8. - №15 – P.839-860. DOI: 10.1111/tra.12176.
- Dale J.M. X-linked spinal muscular atrophy in mice caused by autonomous loss of ATP7A in the motor neuron / L. Hodgkinson et al. // J Pathol. – 2015. – V.2. - №236 – P.241-250. DOI: 10.1002/path.4511.
- Kennerson M.L. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy / M.L. Kennerson et al. // Am J Hum Genet. – 2010. – V.3. - №86 – P.343-352. DOI: 10.1016/j.ajhg.2010.01.027.
- Braiterman L. Apical targeting and Golgi retention signals reside within a 9-amino acid sequence in the copper-ATPase, ATP7B / L. Braiterman et al. // Am J Physiol Gastrointest Liver Physiol. – 2009. – V.2. - №296 – P.433-444. DOI: 10.1152/ajpgi.90489.2008.
- Donoso M. Polarized traffic of LRP1 involves AP1B and SNX17 operating on Y-dependent sorting motifs in different pathways / M. Donoso et al. // Mol Biol Cell. – 2009. – V.1. - №20 – P.481-497. DOI: 10.1091/mbc.e08-08-0805.
- Gromadzka G. Genetic variability in the methylenetetrahydrofolate reductase gene (MTHFR) affects clinical expression of Wilson's disease / G. Gromadzka et al. // J Hepatol. – 2011. – V.4. - №55 – P.913-919. DOI: 10.1016/j.jhep.2011.01.030.
- Weiss K.H. Copper-induced translocation of the Wilson disease protein ATP7B independent of Murr1/COMMD1 and Rab7 / K.H. Weiss et al. // Am J Pathol. – 2008. – V.6. - №173 – P.1783-1794. DOI: 10.2353/ajpath.2008.071134.
- Simon I. Analysis of the human Atox 1 homologue in Wilson patients / I. Simon et al. // World J Gastroenterol. – 2008. – V.15. - №14 – P.2383-2387. DOI: 10.3748/wjg.14.2383.
- Weiss K.H. Genetic analysis of BIRC4/XIAP as a putative modifier gene of Wilson disease / K.H. Weiss et al. // J Inherit Metab Dis. – 2010. - №33 – P.233-240. DOI: 10.1007/s10545-010-9123-5.
- Stättermayer A.F. Hepatic steatosis in Wilson disease--Role of copper and PNPLA3 mutations / A.F. Stättermayer et al. // J Hepatol. – 2015. – V.1. - №63 – P.156-163. DOI: 10.1016/j.jhep.2015.01.034.
- Przybyłkowski A. Polymorphisms of metal transporter genes DMT1 and ATP7A in Wilson's disease / A. Przybyłkowski et al. // J Trace Elem Med Biol. – 2014. – V.1. - №28 – P.8-12. DOI: 10.1016/j.jtemb.2013.08.002.
- Ferenci P. Diagnosis and phenotypic classification of Wilson disease / P. Ferenci et al. // Liver Int. – 2003. – V.3. - №23 – P.139-142. DOI: 10.1034/j.1600-0676.2003.00824.x.
- Nicastro E. Re-evaluation of the diagnostic criteria for Wilson disease in children with mild liver disease / E. Nicastro et al. // Hepatology. – 2010. – V.6. - №52 – P.1948-1956. DOI: 10.1002/hep.23910.
- Ferenci P. Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing / P. Ferenci // Hum Genet. – 2006. – V.2. - №120 – P.151-159. DOI: 10.1007/s00439-006-0202-5.
- Sanger F. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase / F. Sanger et al. // Journal of molecular biology. – 1975. – V.3. - №94 – P.441-448.
- Chang I.J. The genetics of Wilson disease / I.J. Chang et al. // Handb Clin Neurol. – 2017 - №142 – P.19-34. DOI: 10.1016/B978-0-444-63625-6.00003-3.
- Bost M. Molecular analysis of Wilson patients: direct sequencing and MLPA analysis in the ATP7B gene and Atox1 and COMMD1 gene analysis / M. Bost et al. // J Trace Elem Med Biol. – 2012. - №26 – P.97-101. DOI: 10.1016/j.jtemb.2012.04.024.
- Lepori M.B. Mutation analysis of the ATP7B gene in a new group of Wilson's disease patients: contribution to diagnosis / M.B. Lepori et al. // Mol Cell Probes. – 2012. – V.4. - №26 – P.147-150. DOI: 10.1016/j.mcp.2012.03.007.
- Lin C.W. Development of a high-resolution melting method for the screening of Wilson disease-related ATP7B gene mutations / C.W. Lin et al. // Clin Chim Acta. – 2010. – V.17-18. - №411 – P.1223-1231. DOI: 10.1016/j.cca.2010.04.030.
- Mak C.M. Diagnosis of Wilson's disease: a comprehensive review. / C.M. Mak et al. // Crit Rev Clin Lab Sci. – 2008 – V.3. - №45 – P.263-290. DOI: 10.1080/10408360801991055.
- Gomes A. Geographic distribution of ATP7B mutations in Wilson disease / A. Gomes et al. // Ann Hum Biol. – 2016. – V.1. - №43 – P.1-8. DOI: 10.3109/03014460.2015.1051492.
- Mahdieh N. An overview of mutation detection methods in genetic disorders / N. Mahdieh et al. // Iran J Pediatr. – 2013. – V.4. - №23 – P.375-388.
- Arnheim N. Use of pooled DNA samples to detect linkage disequilibrium of polymorphic restriction fragments and human disease: studies of the HLA class II loci / N. Arnheim et al. // Proc Natl Acad Sci USA. – 1985. – V.20. - №82 – 6970-6974. DOI: 10.1073/pnas.82.20.6970.
- Hayashi K. How sensitive is PCR-SSCP / K. Hayashi et al. // Hum Mutat. – 1993. – V.5. - №2 – P.338-346. DOI: 10.1002/humu.1380020503.
- Schouten J.P. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification / J.P. Schouten et al. // Nucleic Acids Res. – 2002. – V.12. - №30 – P.57. DOI: 10.1093/nar/gnf056.
- Newton C.R. Amplification refractory mutation system for prenatal diagnosis and carrier assessment in cystic fibrosis / C.R. Newton et al. // Lancet. – 1989. – V.2. - №30 – P.1481-1483. DOI: 10.1016/s0140-6736(89)92931-0.
- Bathelier C. ARMS test for diagnosis of factor V Leiden mutation and allele frequencies in France / C. Bathelier et al. // Mol Cell Probes. – 1998. – V.2. - №12 – P.121-123. DOI: 10.1006/mcpr.1997.0152.
- Zappu A. Development of TaqMan allelic specific discrimination assay for detection of the most common Sardinian Wilson's disease mutations. Implications for genetic screening / A. Zappu et al. // Mol Cell Probes. – 2010. – V.4. - №24 – 233-235. DOI: 10.1016/j.mcp.2010.01.004.
- Gojová L. Genotyping microarray as a novel approach for the detection of ATP7B gene mutations in patients with Wilson disease / L. Gojová et al. // Clin Genet. – 2008. – V.5. - №73 – P.441-452. DOI: 10.1111/j.1399-0004.2008.00989.x.
- Lu Y. Clinical and molecular characterization of Wilson's disease in China: identification of 14 novel mutations / H. Li et al. // BMC Med Genet. – 2011. – №12 - P.6. DOI: 10.1186/1471-2350-12-6.
- Kumar N. Myelopathy due to copper deficiency following gastrointestinal surgery / N. Kumar et al. // Arch Neurol. – 2003. – V.12. - №60 – P.1782-1785. DOI: 10.1001/archneur.60.12.1782. PMID: 14676057.