Анализ редкого клинического случая: инфантильная форма болезни Помпе с поражением сердца
Анализ редкого клинического случая: инфантильная форма болезни Помпе с поражением сердца
Аннотация
Болезнь Помпе представляет собой сложное генетическое аутосомно-рецессивное заболевание, вызванное мутациями в гене GAA, кодирующий фермент кислую альфа-глюкозидазу, участвующий в расщеплении гликогена лизосомами. В данной статье рассматривается уникальная клиническая картина младенческой формы болезни Помпе, выявленной в возрасте 3 месяцев жизни. Данный разбор клинического случая представляет собой глубокое исследование мультисистемных проблем у ребенка, подчеркивая важность ранней генетической диагностики и ферментозаместительной терапии. Цель исследования — изучить клинические, диагностические и терапевтические особенности инфантильной формы болезни Помпе на примере редкого клинического случая.
1. Введение
Лизосомные болезни накопления — это гетерогенная группа наследственных метаболических заболеваний, характеризующихся прогрессирующей клеточной дисфункцией и системными проявлениями вследствие дефицита лизосомных ферментов и накопления нерасщепленных субстратов. Среди них болезнь Помпе (гликогеноз II типа) — сложное наследственное заболевание, относящееся к группе лизосомных болезней накопления и характеризующееся полисистемным поражением. Особую клиническую значимость представляет младенческая форма заболевания, проявляющаяся выраженной органной дисфункцией и мультисистемным вовлечением, что определяет неблагоприятный прогноз для пациентов . Эпидемиологическая ситуация по болезни Помпе в Российской Федерации недостаточно изучена. Регистрируются лишь отдельные случаи, что затрудняет определение точной частоты встречаемости заболевания. В мировой литературе суммарная частота всех форм болезни Помпе оценивается как 1:40 000 .
Болезнь Помпе представляет собой редкое генетическое расстройство, наследуемое по аутосомно-рецессивному типу. Причиной заболевания являются мутации в гене, отвечающем за синтез фермента кислой альфа-глюкозидазы (GAA), который локализован на участке 17q25 хромосомы . Основным механизмом развития болезни Помпе является патологическое отложение гликогена в различных органах и тканях, при этом наибольшему поражению подвергаются: сердечная мышца, скелетная и гладкая мускулатура. Изначально заболевание классифицировалось как первично-мышечное, однако современные исследования расширили представление о его патогенезе, выявив влияние на эндотелиальные клетки и мотонейроны. Накопление гликогена в центральной нервной системе приводит к развитию каскада патологических процессов, включая нейродегенерацию, васкулопатию и когнитивные расстройства, что подтверждает мультисистемное вовлечение . Дыхательная дисфункция, часто встречающаяся при болезни Помпе, обусловлена комплексным поражением, включающим не только мышечную слабость, но и структурные и функциональные изменения в проксимальных и дистальных дыхательных путях .
Прогрессирующее течение является характерной особенностью болезни Помпе, при этом выделяют инфантильную и взрослую формы, отличающиеся временем манифестации и тяжестью клинических проявлений. Инфантильная форма, дебютирующая в первые месяцы жизни, характеризуется наиболее агрессивным течением и высокой летальностью в течение первого года жизни. К основным клиническим проявлениям инфантильной формы относят: выраженную мышечную слабость, приводящая к генерализованной гипотонии, задержку формирования двигательных навыков, повышенную восприимчивость к респираторным инфекциям, дыхательную недостаточность, кардиомегалию, гипертрофическую кардиомиопатию с развитием сердечной недостаточности, гепатоспленомегалию, а также макроглоссию . В некоторых случаях гипертрофическая кардиомиопатия развивается внутриутробно. Без заместительной ферментной терапии, заболевание быстро прогрессирует, и пациенты обычно умирают на первом году жизни .
К основным методам диагностики, помимо детально собранного анамнеза жизни и заболевания, относят также лабораторные и молекулярно-генетические методы исследования. Особое внимание уделяется наличию случаев болезни Помпе или других генетических заболеваний в семье. Следует уточнить наличие необъяснимых смертей в младенческом возрасте. Определение активности кислой альфа-глюкозидазы в пятнах высушенной крови методом тандемной масс-спектрометрии является эффективным и перспективным подходом для скрининга болезни Помпе, обладающим высокой чувствительностью и специфичностью. При инфантильной форме заболевания активность кислой альфа-глюкозидазы значительно снижается (менее 10% от нормы) . При болезни Помпе наблюдается повышение уровня креатинфосфокиназы (КФК) в крови, что свидетельствует о разрушении мышечных клеток (миоцитолизе), а также повышенное выделение тетрасахарида глюкозы (Glc4) с мочой .
При выявлении сниженной активности кислой альфа-глюкозидазы всем пациентам необходимо проводить молекулярно-генетическое исследование гена GAA. Для обнаружения точечных мутаций в этом гене наиболее точным методом считается прямое секвенирование по Сэнгеру, которое является «золотым стандартом». Next Generation Sequencing (NGS) технология позволяет одновременно секвенировать все экзоны гена GAA, что значительно увеличивает скорость и эффективность анализа . Секвенирование нового поколения является полезным для выявления редких и ранее не описанных мутаций. Существуют различные варианты NGS, включая таргетное секвенирование (анализ только гена GAA) и секвенирование всего экзома, что позволяет одновременно выявлять мутации в гене GAA и других генах, ассоциированных с лизосомными болезнями накопления.
На сегодняшний день основой патогенетической терапии является ферментозаместительная терапия. Первым препаратом, получившим одобрение, стала алглюкозидаза альфа в 2006 году . Клинические исследования демонстрируют, что применение данного препарата у пациентов с болезнью Помпе приводит к существенному увеличению продолжительности жизни, а также к улучшению переносимости физических нагрузок (увеличению дистанции ходьбы) и стабилизации легочной функции, что особенно актуально при сопутствующей легочной недостаточности , . Внутривенное введение алглюкозидазы альфа рекомендуется в дозе 20 мг/кг один раз в две недели .
Информированное согласие на публикацию
От законного представителя пациента получено письменное информированное согласие на публикацию персональной медицинской информации в обезличенной форме.
2. Клиническое наблюдение
Ребенок от 3 беременности, протекавшей на фоне анемии, роды срочные в 39 недель. При рождении: масса тела 4014 грамм, рост 56 см. Пренатально на 32 недели беременности был диагностирован врожденный порок сердца (ВПС). Пациент А. рожден в октябре 2024 года в ФГБУ «НМИЦ акушерства, гинекологии и перинатологии имени академика В.И. Кулакова». Состояние при рождении относительно стабильное, по шкале Апгар 7–8 баллов, ВПС подтвержден, оперативного лечения не требовалось. В период новорожденности обследован в условиях ФГБУ НМИЦ сердечно-сосудистой хирургии имени А.Н.Бакулева. Был выставлен диагноз: ВПС: недостаточность трикуспидального клапана 3 степени, дефект межпредсердной перегородки, открытый артериальный проток, гипоплазия легочной артерии, недостаточность кровообращения 2А степени, функциональный класс 2 по классификации NYHA.
При рождении и в период новорожденности у пациента наблюдалась выраженная генерализованная мышечная гипотония, проявляющаяся общей вялостью, значительно сниженным сопротивлением при пассивных движениях в конечностях, а также слабой способностью удерживать голову. Неврологический статус характеризовался гипорефлексией, включая вялые сухожильные рефлексы, что свидетельствовало о поражении периферического нейромышечного аппарата. Парадоксально, но на фоне общей мышечной слабости отмечалась псевдогипертрофия, преимущественно икроножных мышц, которые при пальпации определялись как плотные, что является характерным признаком для нарушений метаболизма гликогена в мышечной ткани. Трудности с кормлением были обусловлены дисфагией, связанной с мышечной слабостью глотки, гортани и языка. Ребенок быстро уставал при акте сосания, что требовало длительных пауз для восстановления сил. Наряду с этими трудностями, отмечались частые срыгивания, усугубляющие риск аспирации и затрудняющие поддержание нутритивного статуса. Наблюдались эпизоды поверхностного дыхания и периодически возникающая одышка, что было связано как с общей мышечной слабостью, так и с сопутствующим ВПС. Помимо указанных проявлений, при объективном осмотре также отмечалась гепатомегалия.
В связи с отягощенным наследственным анамнезом, объективным осмотром и данными лабораторных методов исследования в условиях лаборатории ФГБУ «НМИЦ акушерства, гинекологии и перинатологии имени академика В.И. Кулакова» в декабре 2024 года выполнено полное секвенирование экзома. Обнаружено 2 вероятно патогенных варианта нуклеотидной последовательности (с.1822С> Т; с.2189+1G> А) в гене GAA.
В феврале 2025 года в условиях лаборатории наследственных болезней обмена веществ ФГБНУ «Медико-Генетический Научный Центр Имени Академика Н.П. Бочкова» выполнена энзимодиагностика: по результатам исследования выявлено снижение активности альфа-глюкозидазы-0,3 мкМ/л.ч при норме 1-25 мкМ/л.ч.
В марте 2025 года в условиях ФГБНУ «МГНЦ» методом массового параллельного секвенирования на приборе Illumina NextSeq проведен полный анализ гена GAA. Выявлены варианты: описанный ранее как патогенный (HGMD_ID CM082738) вариант нуклеотидной последовательности в экзоме 13 гена GAA (сhr17:80112645С> Т) в гетерозиготном состоянии, приводящий к появлению преждевременного терминирующего кодона (NM_000152.5:с.1822С>Т,q.(Arg608*)). Выявлен описанный ранее как патогенный (HGMD_ID CS083933) вариант нуклеотидной последовательности в донорном сайте сплайсинга, интроне 15 гена GAA (сhr17:80113367G>A) в гетерозиготном состоянии, приводящий к нуклеотидной замене (NM_000152.5: с.2189+1 G> A).
Также в марте 2025 года в условиях лаборатории селективного скрининга ФГБНУ «МГНЦ» в рамках научных исследований методом ВЭЖХ-МС/МС проведено определение концентрации Glc4 (тетрасахарид глюкозы) в моче. Концентрация Glc4 составила: 56,91 ммоль/моль креатинина при норме 2,4–16,2 ммоль/моль креатинина.
Важно, что выявленные мутации ранее были описаны в научной литературе, что предполагает потенциальную генотип-фенотипическую корреляцию. Гетерозиготное состояние этих мутаций указывает на носительство и потенциальную семейную генетическую предрасположенность. Данные генетические мутации приводят к значительному снижению функциональной активности фермента альфа-глюкозидазы. В результате этого нарушается процесс расщепления гликогена в лизосомах, и он начинает накапливаться в этих органеллах, вызывая их перегрузку и клеточную дисфункцию. Течение болезни оказало существенное влияние на развитие нервно-мышечной системы пациента. Клинические наблюдения постоянно отмечали мышечную гипотонию, снижение мышечного тонуса и прогрессирующую слабость. У пациента наблюдалась значительная задержка двигательного развития, типичная для раннего начала болезни Помпе.
Наблюдался генетиком с диагнозом: Болезнь Помпе, неонатальная форма, аутосомно-рецессивный тип наследования, с поражением сердца (гипертрофическая кардиомиопатия), ЖКТ (гепатомегалия). Вернувшись домой появилась одышка, сухой кашель, слабость, снижение массы тела. В марте 2025 года были госпитализированы в отделение реанимации и интенсивной терапии ГБУЗ СК «ГДКБ им. Г.К. Филиппского» г. Ставрополь. Проведена телемедицинская консультация с НМИЦ им. В.А. Алмазова: подтвержден диагноз болезни Помпе, обоснована необходимость ферментозаместительной терапии алглюкозидазой альфа по схеме: 20 мг/кг каждые 2 недели.
Для дальнейшего лечения и наблюдения пациент был переведен в ГБУЗ СК КДКБ г. Ставрополь отделение кардиологии и ревматологии, где в дальнейшем проводились лабораторно-инструментальные методы исследования, а также патогенетическая и симптоматическая терапия. Было выполнено рентгенографическое исследование органов грудной клетки, где выявлены признаки сгущения легочного рисунка в проекции S3 правого легкого; рентгенпризнаки резко выраженной кардиомегалии; тень сердца резко расширена в поперечнике, КТИ= 0,71 (рис. 1).
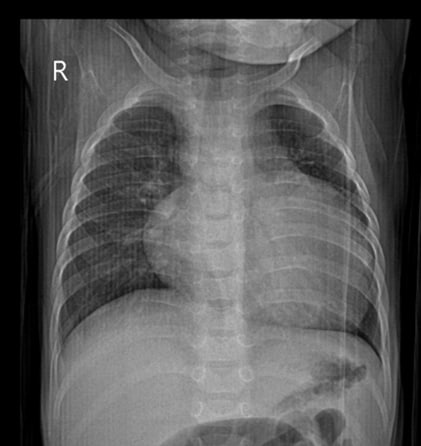
Рисунок 1 - Рентгенография органов грудной клетки на момент госпитализации в ГБУЗ СК КДКБ г. Ставрополь
Последующая ферментозаместительная терапия с июня 2025 года осуществлялась препаратом авалглюкозидаза альфа. Переход на данный препарат, стал возможен благодаря его выделению из Фонда поддержки детей с тяжёлыми жизнеугрожающими и хроническими заболеваниями, в том числе редкими (орфанными) заболеваниями «Круг добра». Это решение было обусловлено как доступностью нового препарата через фонд, так и его улучшенным фармакологическим профилем (увеличенное содержание остатков маннозо-6-фосфата), что обеспечивает более эффективный захват фермента клетками и потенциально лучшую клиническую эффективность. Препарат авалглюкозидаза альфа применялся в его стандартной терапевтической дозировке 40 мг/кг на 1 введение каждые две недели.
На фоне проводимой ферментозаместительной терапии препаратом авалглюкозидаза альфа наблюдалась положительная динамика со стороны сердечно-сосудистой системы, проявляющаяся в улучшении сократительной функции миокарда левого желудочка, о чем свидетельствует увеличение фракции выброса (ФВ). Согласно данным ЭхоКГ, ФВ по Teicholz возросла с 43% (март 2025 года) до 63% (октябрь 2025 года), а ФВ по Simpson — с 45% до 61% за тот же период (рис. 2). Кроме того, отмечалось уменьшение степени легочной гипертензии в период с марта по октябрь 2025 года: систолическое давление в легочной артерии снизилось с 47 мм/Hg до 33 мм/Hg (рис. 3).
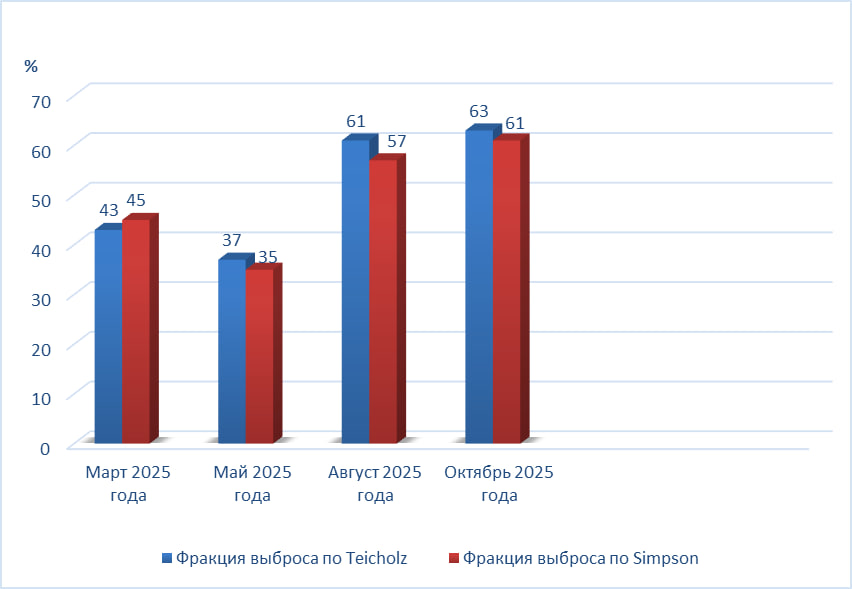
Рисунок 2 - Показатели Эхо-КГ (фракция выброса по Teicholz и Simpson) в динамике

Рисунок 3 - Показатели Эхо-КГ (систолическое давление в легочной артерии) в динамике
Таблица 1 - Биохимические показатели крови в динамике
Дата | АЛТ, е/л; N=0-33 | АСТ, е/л; N=0-37 | КФК, е/л; N=39-308 | КФК - МВ, е/л; N=0-25 | NT-proBNP, пг/мл; N=0-320 |
Апрель 2025 года | 234 | 436 | 1105 | 51 | 10822,0 |
Июнь 2025 года | 160 | 418 | 1167 | 41 | 9833,0 |
Август 2025 года | 89 | 300 | 995 | 41 | 2232,0 |
Октябрь 2025 года | 81 | 324 | 1175 | 29 | 24,7 |
Примечание: АЛТ - аланинаминотрансфераза, АСТ - аспартатаминотрансфераза, КФК - креатинфосфокиназа, КФК - МВ - креатинфосфокиназа-МВ, NT-proBNP - мозговой натрийуретический пептид, N - нормальные значения
3. Заключение
Представленное клиническое наблюдение демонстрирует гетерогенность клинических проявлений инфантильной формы болезни Помпе и необходимость комплексного подхода к диагностике и лечению. Несмотря на наличие ВПС, диагностированного антенатально, заподозрить метаболическое заболевание удалось лишь после рождения. Применение современных методов генетической и биохимической диагностики позволило подтвердить диагноз и своевременно начать ферментозаместительную терапию, что привело к улучшению сердечной функции и метаболического статуса пациента. Однако сохраняющиеся признаки кардиомиопатии и необходимость длительного наблюдения подчеркивают необходимость дальнейших исследований в области патогенеза и лечения болезни Помпе. Крайне важна также доступность ферментозаместительной терапии и мультидисциплинарный подход к ведению таких пациентов.