СИНДРОМ КОРНЕЛИИ ДЕ ЛАНГЕ. КЛИНИЧЕСКИЙ СЛУЧАЙ У РЕБЕНКА ИЗ ДВОЙНИ

СИНДРОМ КОРНЕЛИИ ДЕ ЛАНГЕ. КЛИНИЧЕСКИЙ СЛУЧАЙ У РЕБЕНКА ИЗ ДВОЙНИ
Научная статья
Балакирева Е.А.1, *, Улезько А.А.2, Плетенская С.Р.3, Халаимова О.А.4, Хатамова Х.А.5, Сарычева М.В.6, Пожидаева Т.И.7, Матвиенко Е.В.8, Любова Д.В.9
1 ORCID: 0000-0002-3919-7045;
2 ORCID: 0000-0001-6013-2695;
3 ORCID: 0000-0002-5161-1253;
4 ORCID: 0000-0002-2466-8596;
5 ORCID: 0000-0002-0609-688X;
6 ORCID: 0000-0003-1128-6262;
1-9 Белгородский государственный национальный исследовательский университет, Белгород, Россия
* Корреспондирующий автор (balakireva26[at]mail.ru)
АннотацияПроведен анализ клинического случая ребенка в возрасте четырех лет с диагнозом резидуально-органическое поражение центральной нервной системы на фоне синдрома Корнелии де Ланге, поступившую в отделение реанимации и интенсивной терапии по поводу отсутствия спонтанного дыхания. Девочка из монозиготной дихориальной диамниотической двойни, второй близнец условно здоров. Случай интересен тем, что согласно критериям консенсуса он имеет 11 баллов, а значит относится к классической форме синдрома Корнелии де Ланге, но, учитывая полное отсутствие пороков развития внутренних органов, данный случай занимает пограничное положение, что, по нашим предположениям обуславливает летальный исход больной.
Ключевые слова: синдром Корнелии де Ланге, сепсис, инфекционно-токсический шок, дихориальная диамниотическая двойня.
CORNELIA DE LANGE SYNDROME. A CLINICAL CASE OF A TWIN CHILD.
Research article
Balakireva E.A.1, *, Ulezko A.A.2, Pletenskaya S.R.3, Khalaimova O.A.4, Khatamova Kh.A.5, Sarycheva M.V.6, Pozhidaeva T.I.7, Matvienko E.V.8, Lyubova D.V.9
1 ORCID: 0000-0002-3919-7045;
2 ORCID: 0000-0001-6013-2695;
3 ORCID: 0000-0002-5161-1253;
4 ORCID: 0000-0002-2466-8596;
5 ORCID: 0000-0002-0609-688X;
6 ORCID: 0000-0003-1128-6262;
1-9 Belgorod State National Research University, Belgorod, Russia
* Corresponding author (balakireva26[at]mail.ru)
AbstractThe analysis of a clinical case of a child aged four years with a diagnosis of a residual organic lesion of the central nervous system against the background of Cornelia de Lange syndrome, who was admitted to the intensive care unit for lack of spontaneous breathing, was carried out. A girl from monozygotic dichoric diamniotic twins, the second twin is conditionally healthy. The case is interesting because, according to the consensus criteria, it has 11 points, which means it belongs to the classical form of Cornelia de Lange syndrome, but given the complete absence of malformations of internal organs, this case occupies a borderline position, which, according to our assumptions, causes a fatal outcome of the patient.
Keywords: Cornelia de Lange syndrome, sepsis, infectious toxic shock, dichorial diamniotic twins.
ВведениеСиндром Корнелии де Ланге (CdLS) — мультисистемное расстройство с физическими, когнитивными и поведенческими характеристиками. Названо в честь голландского педиатра Корнелии де Ланге, впервые описавшейданный синдром у двух младенцев в 1933 году [1]. CdLS является клинически и генетически гетерогенным расстройством развития. Клинические особенности включают задержку роста, интеллектуальный дефицит, дефекты конечностей, типичный дисморфизм лица и другие системные поражения. Более глубокое понимание генетической основы CdLS привело к улучшению диагностики и расширению фенотипа. Были идентифицированы мутации в пяти генах (NIPBL, SMC1A, SMC3, RAD21 и HDAC8), всех регуляторах или структурных компонентах когезина. Примерно 60% случаев CdLS связаны с мутациями NIPBL, 5% вызваны мутациями в SMC1A, RAD21 и HDAC8, а один пробанд несет мутацию в SMC3. На сегодняшний день известно 311 Мутаций, вызывающих CdLS [2].
Спектр CdLS связан с молекулярными аномалиями, влияющими на гены, участвующие в регуляции хроматина, чаще всего те, которые связаны с когезивным комплексом. Иначе говоря, синдром Корнелии де Ланге – это когезинопатия [3], [4], [7], [8]. Диагноз CdLS устанавливается у пробанда с наводящими клиническими признаками и/или путем идентификации гетерозиготного патогенного варианта в NIPBL, RAD21, SMC3или BRD4 или гемизиготного патогенного варианта в HDAC8 или SMC1A методом молекулярно-генетического исследования.
В рамках генетического консультирования: NIPBL-CdLS, RAD21-CdLS, SMC3-CdLS и BRD4-CdLS наследуются аутосомно-доминантным образом; HDAC8-CdLS и SMC1A-CdLS наследуются X-сцепленно. Большинство пораженных лиц имеют de novo гетерозиготный патогенный вариант в NIPBL. Менее 1% людей с аутосомно-доминантным CdLS имеют пораженного родителя. Когда родители клинически не больны, риск для сибсов пробанда с CdLS оценивается в 1,5% из-за возможности мозаицизма зародышевой линии. Риск для сибсов мужского пробанда с Х-сцепленным CdLS зависит от статуса матери пробанда; риск для братьев и сестер женщины с X-сцепловым CdLS зависит от статуса матери и отца пробанда. Пренатальное тестирование на беременность с повышенным риском, а также предимплантационное генетическое тестирование возможны для семей, у которых был идентифицирован патогенный вариант [9].
Ряд авторов разработали критерии консенсуса, используя следующие особенности:
Главные признаки (по 2 очка при наличии):
- Синофриз и/или густые брови.
- Короткий нос, вогнутый носовой гребень и/или перевернутый кончик носа.
- Длинный и/или гладкий фильтрум.
- Тонкая верхняя губа и/или опущенные уголки рта.
- Ручная олигодактилия и/или адактилия.
- Врожденная диафрагмальная грыжа.
Дополнительные (по 1 баллу, если они присутствуют):
- Глобальная задержка развития и/или интеллектуальная инвалидность.
- Пренатальная задержка роста.
- Постнатальная задержка роста.
- Микроцефалия (пренатально и/или постнатально).
- Маленькие руки и/или ножки.
- Короткий пятый палец.
- Гирсутизм.
Оценка ≥11 указывает на классический CdLS, если присутствуют по крайней мере три главных признака; оценка 9–10 указывает на неклассическую CdLS, если присутствуют хотя бы два главных признака; оценка ≥4 достаточна для того, чтобы оправдать молекулярное тестирование на CdLS, если присутствует по крайней мере один главный признак; оценка ниже <4 недостаточна для указания на такое тестирование (R2). Оценка ≥11 подтверждает диагноз CdLS независимо от того, можно ли найти патогенный вариант в одном из известных генов [10].
Для оценки частоты фенотипических проявлений синдрома Корнелии де Ланге мы сравнили данные Козловой С.И и соавт. [11] и данные из каталога Мак-Кьюзика [12]. В скобках частота встречаемости обозначена в виде процентов из первого и второго источника соответственно. В том случае, если разница между процентами не превышает 10 %, мы берем усредненное значение. Наблюдаемые признаки: микробрахицефалия (93%, 73%), деформированные ушные раковины (58%), синофриз (99%, 73%), тонкие брови, длинные загнутые ресницы (95% ), маленький нос с открытыми вперед ноздрями (100%, 57%), атрезия хоанн, длинный выступающий фильтрум (94%, 67%), микрогения (97%, 40%), тонкая верхняя губа(94%, 81%), рот в виде полумесяца (94%, 60%), высокое арковидное небо(86%, 45%), большие промежутки между зубами (86%, 38,5%), иногда расщелина неба, акромикрия (93%), ограничение подвижности локтевых суставов (64%), фокомелия и олигодактилия (30%), клинодактилия 5-го пальца (69%), гипертрихоз (78%), мраморность кожного покрова (60%), гипоплазия сосков (55%), слабый высокий голос (74%), судороги(20%). Описаны пороки внутренних органов: врожденные пороки сердца (17%), поликистоз почек, гидронефроз, удвоение или неполный поворот кишечника, пилоростеноз, паховые и диафрагмальные грыжи, крипторхизм (73%), гипоплазия гениталий у мальчиков (57%). Типичны рецидивирующие респираторные инфекции.
Аномалии глаз- миопия, микрокорнеа, астигматизм, атрофия или колобома зрительного нерва, косоглазие.
В рамках фенотипической вариабельности выделяют два варианта синдрома:
- Классический, характеризуется выраженной парентеральной гипоплазией, значительной задержкой физического и умственного развития.
- Доброкачественный, сопровождается аналогичными лицевыми и малыми скелетными аномалиями, пограничной задержкой психомоторного развитии. Крупные пороки развития отсутствуют [11], [12].
В литературе впервые в 1976 г была описана пара монозиготных близнецов женского пола, один из которых страдал данным синдромом. Монозиготность была установлена с помощью исследования антигена группы крови и сывороточного белка, как у близнецов, так и у их матери. Авторы в качестве гипотезы выдвинули два генетических механизма возникновения синдрома Корнелии де Ланге у одной из девочек. Первая — гипотеза хромосомной или митотической нестабильности. Вторая— возможность постзиготной новой мутации [13].
В 2020г. было проведено секвенирование всего экзома трех вьетнамских пациентов, в результате чего были выявлены две известные гетерозиготные мутации c.6697G>A (p.Val2233Met) и c.2602C>T (p.Arg868X), а также новая гетерозиготная мутация c.4504delG (p.Val1502fsX87) в гене NIPBL. Анализ in silico выявленных мутаций выявил возможное повреждающее воздействие на белок NIPBL. Наследственные анализы в семьях пациентов показали, что все мутации являются de novo [14]. Таким образом эти данные расширяют диагностику пациентов с CdLS и спектр мутаций в гене NIPBL, а также доказывают эффективность секвенирования всего экзома для генетического скрининга CdLS.
Цель исследования
Нами представлен клинический случай ребенка с синдромом Корнелии де Ланге, объективные внешние признаки которого соответствуют классической форме заболевания, в то время как внутренние органы полностью сохранны.
Методы и принципы исследования
Проведен анализ клинического случая ребенка 4 лет с синдромом Корнели де Ланге, поступившего в связи с затруднением дыхания, вызванного аспирацией рвотных масс в отделение реанимации и интенсивной терапии (ОРИТ) ОГБУЗ «Детская областная клиническая больница» г. Белгорода.
Клинический разбор: больная Ш., (на рис. 1), с характерными фенотипическими признаками: недоразвиты проксимальные отделы конечностей, «ластовидные» кисти, уменьшено количество пальцев на кистях рук, густые сросшиеся брови, короткий нос, вогнутый носовой гребень, опущенные уголки рта, длинные загнутые ресницы, гирсутизм.
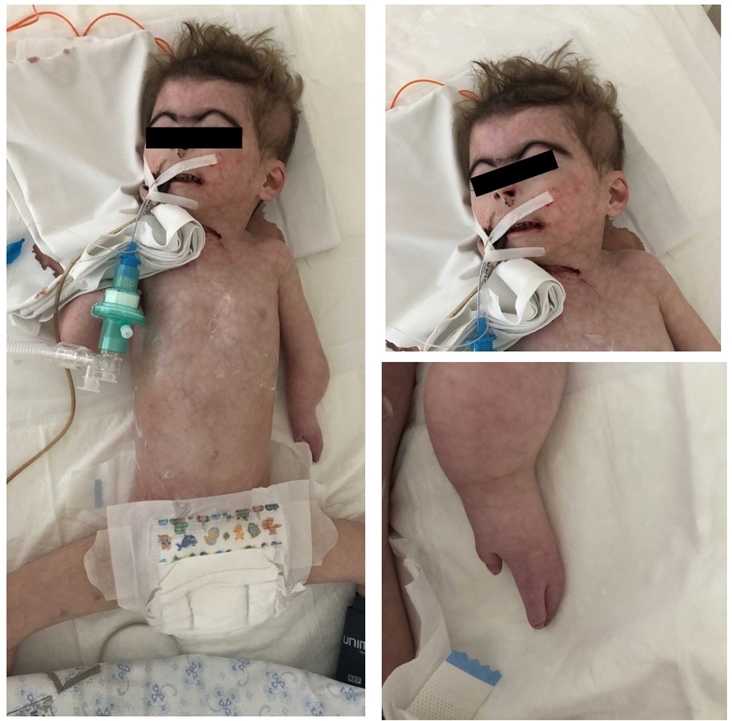
Рис. 1 – Внешний вид больной Ш. с синдромом Корнелии де Ланге
Примечание: собственное клиническое наблюдение
Основной диагноз: резидуально-органическое поражение центральной нервной системы на фоне синдрома Корнелии де Ланге. Глубокая умственная отсталость. Иммунодефицитное состояние, врожденная срединная расщелина твердого неба, врожденная катаракта, миопия слабой степени, ангиопатия сетчатки обоих глаз, малые аномалии развития сердца (открытое овальное окно), дополнительная хорда левого желудочка, кардиопатия, синусовая аритмия, двухсторонняя кондуктивная тугоухость 1-2 ст., анемия децифитная 1 ст., белково-энергетическая недостаточность 3-ей степени. Осложнение основного диагноза: спастический тетрапарез. GMFCS IV уровень, судорожный синдром (медикаментозная ремиссия), задержка статико-моторного и психоречевого развития. Нарушение цикличности сна и бодрствования.
Из анамнеза известно, что ребенок от II беременности (1 беременность от другого брака - здоровый ребенок (13 лет)), дихориальная диамниотическая двойня, протекавшей на фоне угрозы прерывания на сроке 20,23,24 недель, внутриутробной инфекции. Родилась недоношенная в результате оперативных родов на сроке 35 недель. Вес при рождении 1210 г, рост 37 см, окр. головы 26 см. Оценка по шкале Апгар 6-7. Состояние в остром периоде адаптации тяжелое за счет незрелости, врожденных пороков развития, дыхательной недостаточности 2 степени (ДН 2).
Находилась в отделении патологии новорожденных и недоношенных детей перинатального центра на протяжении двух месяцев от момента рождения, после чего была переведена в педиатрическое отделение ОГКУЗ «Белгородский дом ребенка специализированный» со следующим диагнозом: синдром Корнелии де Ланге, врожденная двухстороння катаракта, глаукома, расщелина неба, аномалии развития верхних конечностей, МАРС - открытое овальное окно, железодефицитная анемия 1 ст, задержка внутриутробного развития (ЗВУР) по гипопластическому типу 3 ст.
Вторая девочка из дихориальной диамниотической двойни условно здорова, проживает с родителями. С целью уточнения мутации de novo родителям было предложено провести секвенирование всего экзома на наличие гетерозиготных мутаций в гене NIPBL.
Ребенок заболел остро - стонущее дыхание, температура 39 С, сатурация 80, беспокойство, по тяжести состояния был госпитализирован. После проведения неотложных мероприятий состояние стабилизировалось, но незначительно, поскольку через 8 часов появилось кофейное отделяемое из ротовой полости, напряженность мышц передней брюшной стенки, болезненность живота. В связи с интеллектуальным дефицитом и возрастом ребенка (нервно-психическое развитие соответствует 5 месяцам) точно определить локализацию инфекционного очага не представлялось возможным. В процессе обследования, учитывая результаты клинических, биохимических, бактериологических анализов, а также консультаций узких специалистов- диагноз был изменен. Диагноз, установленный в ОРИТ: сепсис, перитонит, парез кишечника, синдром полиорганной недостаточности, ДВС-синдром, отек мозга, кома 3, иммунодефицитное состояние на фоне синдрома Корнелии де Ланге, спастический тетрапарез GMFCS уровень тяжелой степени тяжести.
Несмотря на проводимую в ОРИТ комплексную терапию (АИВЛ через ЭТТ, инфузионная терапия, антибиотикотерапия, глюкокортикостероидная терапия, гемостатическая терапия, симптоматическая терапия) состояние ребенка прогрессивно ухудшалось, что в конечном итоге привело к летальному исходу в возрасте 4 лет. Смерть ребенка наступила от остро развившегося, возникшего на фоне токсикоза инфекционно-токсического шока с отеком легких, отеком вещества головного мозга с вклинением миндалин мозжечка в большое затылочное отверстие, с развитием ДВС-синдрома с сосудистыми нарушениями в микроциркуляторного русла внутренних органов и головного мозга, очаговыми кровоизлияниями в пульпу селезенки, мелкоточечными кровоизлияниями под капсулу вилочковой железы и надпочечников. Очаговый некроз эпителия канальцев проксимальных канальцев почек.
В ходе проведения патологоанатомического вскрытия выявлен первичный инфекционный очаг - острая кишечная бактериальная инфекция в виде распространенного язвенно-некротического воспаления преимущественно в толстом кишечнике с развитием диффузной гангрены кишечника и разлитого калового перитонита.
Результаты и обсуждениеФоновое иммунодефицитное состояние, характерное для CdLS, в сочетании с гипоплазией вилочковой железы (3 гр при возрастной норме 23 гр) и антибиотикорезистентностью обусловливает стремительное течение инфекционного процесса с развитием сепсиса на фоне антибиотикорезистентности (амикацин, цефоперазон), быстрое прогрессирование полиорганной недостаточности, что и привело к летальному исходу.
Заключение
Случай интересен тем, что:
- Пробанд является монозиготным дихориальным диамниотическим близнецом, имеет здоровую сестру, что подтверждает мутацию de novo.
- Врожденный иммунодефицит в сочетании с частыми острыми инфекциями, характерными для пациентов с CdLS, в данном клиническом случае мог быть причиной молниеносного фатального течения заболевания.
- С учетом наличия 11 диагностических баллов мы должны отнести данный клинический пример картину к классической форме CdLS, но полное отсутствие пороков развития внутренних органов не дает нам этого сделать и ставит данный случай в пограничное положение.
| Конфликт интересов Не указан. | Conflict of Interest None declared. |
Список литературы / References
- De Lange, C. Sur un type nouveau de degenerescence (typus Amsterlodamensis) / C. De Lange // Med. Enfants36, 713–719 (1933).
- Mannini L. Mutation spectrum and genotype-phenotype correlation in Cornelia de Lange syndrome / L. Mannini, Cucco, V. Quarantotti et al. // Hum Mutat. 2013 Dec;34(12):1589-96. DOI: 10.1002/humu.22430.
- Krantz, I. D. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogasterNipped-B / I. D. Krantz et al. // Genet.36, 631–635 (2004).
- Tonkin, E. T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome / E. T. Tonkin, T. J. Wang, S. Lisgo et al. // Genet.36, 636–641 (2004).
- Musio, A. X-Linked Cornelia de Lange syndrome owing to SMC1L1 mutations / A. Musio et al. // Genet.38, 528–530 (2006).
- Deardorff, M. A. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of Cornelia de Lange syndrome with predominant mental retardation / M. A. Deardorff et al. // J. Hum. Genet.80, 485–494 (2007).
- Deardorff, M. A. RAD21 mutations cause a human cohesinopathy / M. A. Deardorff et al. // J. Hum. Genet.90, 1014–1027 (2012).
- Olley, G. BRD4 interacts with NIPBL and BRD4 is mutated in a Cornelia de Lange-like syndrome / G. Olley et al. // Genet.50, 329–332 (2018).
- Deardorff M.A. Cornelia de Lange Syndrome. 2005 Sep 16 [updated 2020 Oct 15] / M.A. Deardorff, S.E. Noon, D. Krantz // GeneReviews. Seattle (WA): University of Washington, Seattle; 1993–2021. PMID: 20301283.
- Kline, A.D. Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement / D. Kline, J.F. Moss, A. Selicorni et al.// Nat Rev Genet19, 649–666 (2018). DOI: 10.1038/s41576-018-0031-0
- Козлова С.И. Наследственные синдромы и медико-генетическое консультирование. Атлас-справочник / С.И. Козлова, И.С. Демикова, Е.С.Семанова. Изд.2-е дополн.- М.:Практика, 1996- 416 стр. ISBN 5-88D01-D08-2
- OMIM®- Online Mendelian Inheritance in Man. [Electronic resource]. URL: https://clck.ru/amJJJ (accessed 09.11.2021)
- Carakushannsky G. The de Lange syndrome in one of twins / G. Carakushannsky, C. Berthier // J Med Genet. 1976 Oct;13(5):404-6. Doi: 10.1136/jmg.13.5.404. PMID: 1034016; PMCID: PMC1013450
- Thanh D.C. De novo NIPBLMutations in Vietnamese Patients with Cornelia de Lange Syndrome / D.C. Thanh, C.T.B. Ngoc, N.L. Nguyen et al. // Medicina (Kaunas). 2020 Feb 14;56(2):76. DOI: 10.3390/medicina56020076. PMID: 32074972; PMCID: PMC7073647.
Список литературы на английском языке / References in English
- De Lange, C. Sur un type nouveau de degenerescence (typus Amsterlodamensis) [On a new type of degenerescence (typus Amsterlodamensis)] / C. De Lange // Med. Enfants36, 713–719. 1933. [in French]
- Mannini L. Mutation spectrum and genotype-phenotype correlation in Cornelia de Lange syndrome / L. Mannini, Cucco, V. Quarantotti et al. // Hum Mutat. 2013 Dec;34(12):1589-96. DOI: 10.1002/humu.22430.
- Krantz, I. D. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogasterNipped-B / I. D. Krantz et al. // Genet.36, 631–635. 2004.
- Tonkin, E. T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome / E. T. Tonkin, T. J. Wang, S. Lisgo et al. // Genet.36, 636–641 (2004).
- Musio, A. X-Linked Cornelia de Lange syndrome owing to SMC1L1 mutations / A. Musio et al. // Genet.38, 528–530 (2006).
- Deardorff, M. A. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of Cornelia de Lange syndrome with predominant mental retardation / M. A. Deardorff et al. // J. Hum. Genet.80, 485–494 (2007).
- Deardorff, M. A. RAD21 mutations cause a human cohesinopathy / M. A. Deardorff et al. // J. Hum. Genet.90, 1014–1027 (2012).
- Olley, G. BRD4 interacts with NIPBL and BRD4 is mutated in a Cornelia de Lange-like syndrome / G. Olley et al. // Genet.50, 329–332 (2018).
- Deardorff M.A. Cornelia de Lange Syndrome. 2005 Sep 16 [updated 2020 Oct 15] / M.A. Deardorff, S.E. Noon, D. Krantz // GeneReviews. Seattle (WA): University of Washington, Seattle; 1993–2021. PMID: 20301283.
- Kline, A.D. Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement / D. Kline, J.F. Moss, A. Selicorni et al.// Nat Rev Genet19, 649–666 (2018). DOI: 10.1038/s41576-018-0031-0
- Kozlova S.I. Nasledstvennye sindromy i mediko-geneticheskoe konsul'tirovanie. Atlas-spravochnik [Hereditary syndromes and medical and genetic counseling. Atlas-handbook] / S.I. Kozlova, I.S. Demikova, E.S.Semanova. Ed.2nd supplement- M.:Praktika, 1996- 416 pages ISBN 5-88D01-D08-2 [in Russian]
- OMIM®- Online Mendelian Inheritance in Man. [Electronic resource]. URL: https://clck.ru/amJJJ (accessed 09.11.2021)
- Carakushannsky G. The de Lange syndrome in one of twins / G. Carakushannsky, C. Berthier // J Med Genet. 1976 Oct;13(5):404-6. Doi: 10.1136/jmg.13.5.404. PMID: 1034016; PMCID: PMC1013450
- Thanh D.C. De novo NIPBLMutations in Vietnamese Patients with Cornelia de Lange Syndrome / D.C. Thanh, C.T.B. Ngoc, N.L. Nguyen et al. // Medicina (Kaunas). 2020 Feb 14;56(2):76. DOI: 10.3390/medicina56020076. PMID: 32074972; PMCID: PMC7073647.