QSAR МОДЕЛИРОВАНИЕ ИНГИБИТОРНОЙ АКТИВНОСТИ КОНЪЮГАТОВγ-КАРБОЛИНОВ И ФЕНОТИАЗИНА ПО ОТНОШЕНИЮ К БУТИРИЛХОЛИНЭСТЕРАЗЕ
QSAR моделирование ингибиторной активности конъюгатовγ-карболинов и фенотиазина по отношению к бутирилхолинэстеразе
Научная статья
Раздольский А.Н.1, *, Казаченко В.П.2, Страхова Н.Н.3, Раевская О.Е.4, Григорьев В.Ю.5
1 ORCID: 0000-0002-3389-4659;
2 ORCID: 0000-0003-1424-1895;
3 ORCID: 0000-0002-6593-2218;
5 ORCID: 0000-0002-5288-3242;
1, 2, 3, 4, 5 Институт физиологически активных веществ РАН, Черноголовка, Россия
* Корреспондирующий автор (rasd[at]ipac.ac.ru)
АннотацияЦелью данного исследования являлось создание QSAR (Количественная связь «структура – активность») моделей ингибиторной активности конъюгатов γ-карболинов и фенотиазина по отношению к бутирилхолинэстеразе (BChE). Эти соединения выступают в качестве селективных ингибиторов BChE и одновременно блокаторами NMDA рецепторов. Поиск и проектирование мультитаргетных соединений, действующих одновременно на несколько молекулярных мишеней, является новым направлением при разработке препаратов для лечения нейродегенеративных заболеваний (болезнь Альцгеймера, болезнь Паркинсона и др.). Для всех молекул был проведен полный конформационный анализ на основе программы CacheWorksystemPro. В качестве дескрипторов пространственного строения молекул применяли интегралы спектров межатомных внутримолекулярных взаимодействий, которые рассчитывали с помощью программы MOLTRA. Для QSAR моделирования использовали метод множественной линейной регрессии. Полученные регрессионные уравнения имеют удовлетворительные статистические характеристики и соответствуют принципам OECD. В дальнейшем они могут быть использованы в качестве фильтров при оптимизации свойств соединений и найти применение в молекулярном виртуальном скрининге.
Ключевые слова: QSAR, мультитаргетные соединения, конъюгаты γ-карболинов и фенотиазина, BChE, MOLTRA.
QSAR Modeling of Inhibitory Activity of γ-Carboline and Phenothiazine Conjugates towards Butyrylcholinesterase
Research article
Razdolsky A.N.1, *, Kazachenko V.P.2, Strakhova N.N.3, Raevskaya O.E.4, Grigoryev V.Yu.5
1 ORCID: 0000-0002-3389-4659;
2 ORCID: 0000-0003-1424-1895;
3 ORCID: 0000-0002-6593-2218;
5 ORCID: 0000-0002-5288-3242;
1, 2, 3, 4, 5 Institute of Physiologically Active Substances of the Russian Academy of Sciences, Chernogolovka, Russia
* Corresponding author (rasd[at]ipac.ac.ru)
AbstractThe purpose of this study was to create a QSAR (Quantitative Structure-Activity Relationship) models of the inhibitory activity of γ-carboline and phenothiazine conjugates towards butyrylcholinesterase (BChE). These compounds act as selective BChE inhibitors and NMDA receptor blockers at the same time. The search and design of multi-target compounds acting simultaneously on several molecular targets is a new trajectory in the development of drugs for the treatment of neurodegenerative diseases (Alzheimer's disease, Parkinson's disease, etc.). A complete conformational analysis was performed for all molecules in Cache Worksystem Pro. Integrals of the spectra of interatomic intramolecular interactions were used as descriptors of the spatial structure of molecules, which were calculated using MOLTRA. For QSAR modeling, the method of multiple linear regression was used. The obtained regression equations have satisfactory statistical characteristics and comply with the OECD principles. In the future, they can be used as filters to optimize the properties of compounds and find application in molecular virtual screening.
Keywords: QSAR, multitargeted compounds, conjugates of γ-carbolines and phenothiazine, BChE, MOLTRA. ВведениеОдной из глобальных проблем современного мира является необходимость борьбы с нейродегенеративными заболеваниями, такими как болезнь Альцгеймера (БА), болезнь Паркинсона и др. В 2020 году, например, в США проживало около 5.8 миллионов человек с БА [1] и общие расходы на здравоохранение достигали 305 миллиардов долларов [2]. В настоящее время лекарств для БА не разработано, а в медицинской практике для симптоматического лечения используется 4 препарата: донепезил, галантамин, ривастигмин и мемантин [3].
Болезнь Альцгеймера является многофакторным заболеванием [4], в связи с чем возникло новое направление при разработке лекарств для БА: проектирование мультитаргетных соединений [5], действующих одновременно на несколько молекулярных мишеней. Учитывая, что донепезил, галантамин и ривастигмин являются ингибиторами холинэстераз, а мемантин – блокатор NMDA рецепторов [3], привлекательной является идея сконструировать соединения, действующие на две вышеперечисленные мишени. Одними из таких соединений являются конъюгаты γ-карболинов и фенотиазина [6], выступающие в качестве селективных ингибиторов бутирилхолинэстеразы (BChE) и блокаторов NMDA рецепторов.
Нужно отметить, что процесс разработки нового лекарства является сложным, требует больших материальных и временных затрат, связан с необходимостью использования междисциплинарной экспертизы и инновационных подходов. Для того чтобы вывести новое лекарственное средство на рынок требуется до 13.5 лет и 1.8 миллиардов долларов [7]. В связи с этим с целью снижения расходов оправданным является использование расчетных методов и подходов, в частности, QSAR (Количественная связь «структура – активность») моделирования. В литературе описаны случаи успешного применения QSAR при поиске новых соединений-лидеров и оптимизации их свойств [8].
Целью настоящей работы является создание QSAR моделей ингибиторной активности конъюгатов γ-карболинов и фенотиазина по отношению к BChE.
Материалы и методы
Обучающая выборка из 11 соединений (рисунок 1, таблица 1) была сформирована на основе работы [6], в которой представлены данные по ингибированию бутирилхолинэстеразы (BChE, EC 3.1.1.8 из лошадиной сыворотки). В качестве меры ингибирующей активности использовали log(1/IC50), где IC50 (мкМ) – концентрация вещества, вызывающая 50% ингибирование BChE.
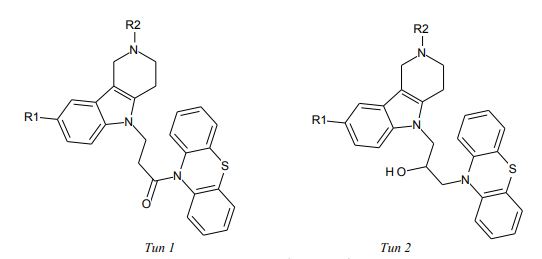
Рис. 1 – Конъюгаты γ-карболинов и фенотиазина
Таблица 1 – Ингибиторная активность конъюгатов γ-карболинов и фенотиазина
| Соединение | Тип | R1 | R2 | IC50, мкМ | log(1/IC50) |
| 1 | 1 | CH3 | CH3 | 62,6 ± 4,3 | -1,80 |
| 2 | 1 | CH3 | С2H5 | 2,04 ± 0,55 | -0,31 |
| 3 | 1 | F | CH3 | 1,79 ± 0,28 | -0,25 |
| 4 | 1 | H | CH3 | 1,07 ± 0,12 | -0,03 |
| 5 | 1 | H | С2H5 | 0,52 ± 0,01 | 0,28 |
| 6 | 1 | F | С2H5 | 0,58 ± 0,06 | 0,24 |
| 7 | 1 | С2H5 | CH3 | 1,36 ± 0,06 | -0,13 |
| 8 | 1 | i-С3H7 | CH3 | 2,79 ± 0,09 | -0,44 |
| 9 | 2 | CH3 | CH3 | 11,7 ± 0,4 | -1,07 |
| 10 | 2 | F | С2H5 | 2,01 ± 0,04 | -0,30 |
| 11 | 2 | CH3O | CH3 | 0,39 ± 0,02 | 0,40 |
Для каждой молекулы был проведен полный конформационный анализ с использованием программы CacheWorksystemPro 6.0 [9]. При этом применяли метод молекулярной механики в параметризации MM2 с порогом выхода из итерационного процесса минимизации энергии в 0.01 ккал/моль. Пространственные структуры с минимальной энергией считали наиболее вероятными.
В качестве дескрипторов пространственного строения молекул использовали интегралы спектров межатомных внутримолекулярных взаимодействий (СМВВ) [10], [11]. Пример расчета СМВВ представлен на рисунке 2. Каждый дескриптор представлял собой площадь прямоугольника с основанием 0.2 ангстрема и вертикалью (ось ординат) равной интенсивности в данном месте спектра СМВВ. Диапазон межатомных расстояний составлял от 0 до 20 ангстрем с шагом 0.2 ангстрема, что приводило к появлению 20/0.2=100 дескрипторов. При этом учитывали парные атом - атомные взаимодействия с участием:
1) Н-доноров (DDF);
2) Н-акцепторов (AAF);
3) Н-доноров и Н-акцепторов (DAF);
4) положительно заряженных атомов (Q++);
5) отрицательно заряженных атомов (Q--);
6) положительно и отрицательно заряженных атомов (Q+-) а также стерическое (ван-дер-ваальсовое) взаимодействие атомов (VDW).
Таким образом, всего для описания структуры каждого соединения использовали 7*100=700 дескрипторов.
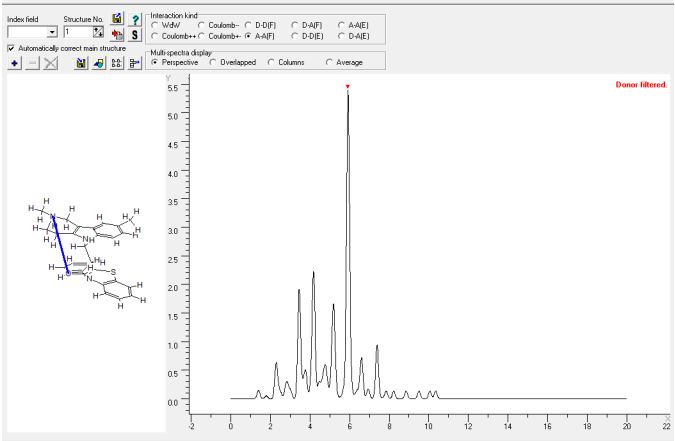
Рис. 2 – Спектр межатомных внутримолекулярных AAF взаимодействий для соединения 1
С целью уменьшения размерности пространства переменных был проведен отбор дескрипторов в каждой группе и общей группе путем анализа корреляционной матрицы с использованием итерационной процедуры, которая состояла из ряда шагов:
1) выбор наиболее информативного дескриптора с максимальной величиной коэффициента вариации;
2) формирование кластера родственных дескрипторов, имеющих коэффициент корреляции с выбранным дескриптором >=0.9;
3) удаление из корреляционной матрицы кластера за исключением выбранного дескриптора;
4) повторение шагов 1-3 до остановки процедуры.
В результате удалось сократить размерность пространства в зависимости от группы до 6-166 дескрипторов.
Для QSAR моделирования использовали компьютерную программу SVD [12], в которой реализован метод множественной линейной регрессии (MLR). Из-за малой величины обучающей выборки применяли только внутреннее тестирование на основе кросс-валидации с выбором по пять (10 итераций). В качестве статистических характеристик моделей использовали: n – число соединений; R2 – коэффициент линейной корреляции; s – стандартное отклонение; FIT – модифицированный критерий Фишера [13]; R2p – рандомизационный параметр [14]. Формирование QSAR моделей проводили с помощью полного перебора комбинаций из 1-2 дескрипторов. Отбор лучших моделей осуществляли на основе статистики FIT. Для оценки области применимости (AD) использовали интервальный метод [15].
Результаты и обсуждение
На первом этапе исследования были получены регрессионные уравнения на основе отдельных групп дескрипторов. Удовлетворительными статистическими характеристиками обладали две представленные ниже модели.
AAF:
| log(1/IC50) = 0.24(±0.22) - 10.1(±2.2) D(5.0-5.2) + 13.0(±2.4) D(7.6-7.8) n=11; R2=0.802; s=0.27; FIT=2.17; R2cv=0.594; scv=0.39; FITcv=0.78; R2p=0.691 AD: D(5.0-5.2) = 0.049÷0.200; D(7.6-7.8) = 0.004÷0.147 | (1) |
VDW:
| log(1/IC50) = -0.54(±0.59) + 0.293(±0.087) D(3.4-3.6) – 0.191(±0.040) D(4.6-4.8) n=11; R2=0.742; s=0.31; FIT=1.53; R2cv=0.517; scv=0.42; FITcv=0.57; R2p=0.658 AD: D(3.4-3.6) = 6.41÷12.29; D(4.6-4.8) =6.50÷19.70 | (2) |
На следующем этапе исследования для генерации QSAR моделей были использованы все имеющиеся в нашем распоряжении 700 дескрипторов СМВВ. После процедуры отбора осталось 166 дескрипторов. В результате был получен ряд регрессионных уравнений, из которых три лучших представлены ниже.
Q-- + Q++:
| log(1/IC50) = -0.27(±0.20) - 118(±17) D(5.8-6.0) + 414(±76) D(6.2-6.4) n=11; R2=0.870; s=0.22; FIT=3.58; R2cv=0.787; scv=0.28; FITcv=1.97; R2p=0.817 AD: D(5.8-6.0) = 0.0031÷0.0200; D(6.2-6.4) = 0.0016÷0.0055 | (3) |
AAF + Q++:
| log(1/IC50) = -1.38(±0.19) – 87.1(±12.9) D(10.0-10.2) + 368(±51) D(4.8-5.0) n=11; R2=0.899; s=0.19; FIT=4.74; R2cv=0.741; scv=0.31; FITcv=1.52; R2p=0.809 AD: D(10.0-10.2) = 0.000÷0.019; D(4.8-5.0) = 0.0019÷0.0070 | (4) |
VDW + Q--:
| log(1/IC50) = -0.33(±0.31) + 4.31(±0.75) D(0.8-1.0) - 278(±40) D(6.0-6.2) n=11; R2=0.867; s=0.22; FIT=3.47; R2cv=0.740; scv=0.31; FITcv=1.51; R2p=0.807 AD: D(0.8-1.0) = 0.269÷0.664; D(6.0-6.2) = 0.0039÷0.0120 | (5) |
На рисунке 3 представлен типичный вид зависимости рассчитанных значений активности от экспериментальных величин. Следует подчеркнуть, что все разработанные QSAR модели удовлетворяют минимальным требованиям, которые к ним предъявляются [16], в частности, R2> 0.6; R2cv> 0.5, R2p> 0.5. Они также вполне соответствуют OECD принципам, связанным с QSAR валидацией [17]:
1) имеется определенная величина активности;
2) однозначный алгоритм расчета;
3) установлена область применимости;
4) приведены соответствующие меры согласия, устойчивости и предсказуемости;
5) возможна механистическая интерпретация.
При этом рассчитанная активность является продуктом баланса между двумя вкладами с противоположными знаками. Расстояния в дескрипторах СМВВ варьируют от 0.8 Å до 10.2 Å. Основная масса дескрипторов связана с расстояниями, превышающими сумму атомных Ван-дер-Ваальсовых радиусов (Å) (RC=1.70, RN=1.55, RO=1.52 [18]), что соответствует области несвязанных атомов.
Важно отметить, что модели (1)-(5) включают в свой состав дескрипторы, которые относятся к четырем группам: AAF, VDW, Q++ и Q--, что отражает вклад водородной связи, стерических и кулоновских взаимодействий в формирование активности. Полученный результат вполне соответствует результатам докинга соединений 6 (IC50=0.58) и 11 (IC50=0.39) в активный центр BChE [6]. Согласно данным этой работы основными видами взаимодействия являются образование Н-связи между гидроксильной группой и оксианионным центром BChE, а также π-π стэкинг между индольным кольцом Trp82 и фенотиазиновым фрагментом. Дополнительно наблюдается слабое π-π взаимодействие между γ-карболиновым фрагментом и Phe329.
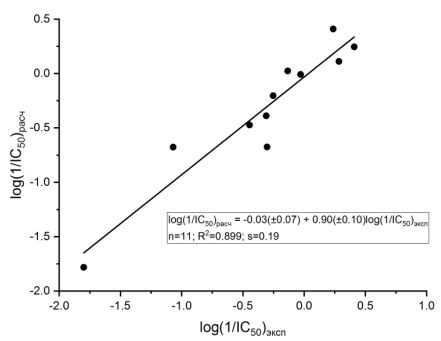
Рис. 3 – Зависимость между экспериментальными и рассчитанными значениями ингибиторной активности соединений 1-11 (модель 4)
Заключение
С использованием метода множественной линейной регрессии сконструированы QSAR модели ингибиторной активности конъюгатов γ-карболинов и фенотиазина. Полученные регрессионные уравнения имеют удовлетворительные статистические характеристики и соответствуют принципам OECD. В дальнейшем они могут быть использованы в качестве фильтров при оптимизации свойств соединений и найти применение в молекулярном виртуальном скрининге.
| Финансирование Эта работа выполнена в рамках Государственного задания ИФАВ РАН 2021 года (тема № 075-00741-22-00 FFSN-2021-0004). | Funding This work was supported by the budget of the Institute of Physiologically Active Compounds of the Russian Academy of Sciences (IPAC RAS) Targets – 2021 (topic No. 075-00741-22-00 FFSN-2021-0004). |
| Конфликт интересов Не указан. | Conflict of Interest None declared. |
- Matthews K.A. Racial and ethnic estimates of Alzheimer’s disease and related dementias in the United States (2015–2060) in adults aged≥65 years / K.A. Matthews, W. Xu, A.H. Gaglioti et al. // Alzheimer’s & Dementia. -2018. - Vol. 15. - P. 17-24. DOI: 10.1016/j.jalz.2018.06.3063
- Wong W. Economic burden of Alzheimer disease and managed care considerations / W. Wong // Am. J. Manag. Care. 2020 Aug;26(8 Suppl): P. 177-183. DOI: 10.37765/ajmc.2020.88482.
- Grossberg G.T. Present algorithms and future treatments for Alzheimer's disease / G.T. Grossberg, G. Tong, A.D. Burke et al. // J. Alzheimer's Dis. - 2019. - Vol. 67. - P. 1157–1171. DOI: 10.3233/JAD-180903
- Carreiras M.C. The multifactorial nature of Alzheimer's disease for developing potential therapeutics / M.C. Carreiras, E. Mendes, M.J. Perry et al. // Curr Top Med Chem.- 2013.- Vol . 13(15). - P. 1745-1770. DOI: 10.2174/15680266113139990135
- Dias K.S. Multi-Target Directed Drugs: A Modern Approach for Design of New Drugs for the treatment of Alzheimer’s Disease / K.S. Dias, C. Viegas // Current Neuropharmacology. - 2014. -Vol. 12.- P. 239-255. DOI: 10.2174/1570159X1203140511153200
- Makhaeva G.F. Conjugates of γ-carbolines and phenothiazine as new selective inhibitors of butyrylcholinesterase and blockers of NMDA receptors for Alzheimer Disease / G.F. Makhaeva, S.V. Lushchekina, N.P. Boltneva et al. // Sci. Rep. - 2015 - №5. - P. 13164. DOI: 10.1038/srep13164.
- Kapetanovic I.M. Overview of Current Drug Discovery and Development with an Eye towards the Future / I.M. Kapetanovic // Drug Discovery and Development - Present and Future [Internet]. London: IntechOpen; 2011.[Electronic resource]. URL: https://www.intechopen.com/chapters/25267(accessed 12.02. 2022)
- Neves B.J. QSAR-Based Virtual Screening: Advances and Applications in Drug Discovery / B.J. Neves, R.C. Braga, C.C. Melo-Filho et al. // Front. Pharmacol. 2018. - 9:1275.DOI: 10.3389/fphar.2018.01275
- Cache Worksystem Pro. [Electronic resource]. URL: http://www.cacheresearch.com. (accessed 12.02. 2022)
- Novikov V.P. Representation of molecular structure in the form of a spectrum of interatomic distances for a study of the relationship of structure to biological activity / V.P. Novikov, O.A. Raevskii // Pharm. Chem. J. - 1982. - Vol. 16(5) - P. 379–386.DOI: 10.1007/BF00762059.
- Trepalin S.V. Software package for computer-aided design of effective physiologically active compounds, based on two-dimentional and three-dimensional physicochemical descriptors / S.V. Trepalin, A.N. Razdolskii, O.A. Raevskii // Pharm. Chem. J.- 2000. – Vol. 34(12). - P. 650-653. DOI: 10.1023/A:1010499601434.
- SVD. [Electronic resource]. URL: https://www.imsl.com(accessed 12.02. 2022)
- Kubinyi H. Variable selection in QSAR studies. I. An evolutionary algorithm / H. Kubinyi // Quant. Struct.-Act. Relat.- 1994. - Vol. 13. - P. 285-294.
- Mitra I. Exploring quantitative structure–activity relationship studies of antioxidant phenolic compounds obtained from traditional Chinese medicinal plants / I. Mitra, A. Saha, K. Roy // Mol. Simul. -2010.- Vol. 36. - P. 1067-1079.
- Roy K. On a simple approach for determining applicability domain of QSAR models / K. Roy, S. Kar, P. Ambure // Chemom. Intell. Lab. Syst. - 2015. - Vol. 145. - P. 22-29. DOI: 10.1016/j.chemolab.2015.04.013
- Kiralj R. Basic Validation Procedures for Regression Models in QSAR and QSPR Studies: Theory and Application / R. Kiralj, M.M.C. Ferreira // J. Braz. Chem. Soc. -2009.-Vol. 20(4). – P. 770-787. DOI: 10.1590/S0103-50532009000400021
- Guidance Document on the Validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models, OECD Series on Testing and Assessment - 2014. - No. 69. OECD Publishing, Paris.DOI: 10.1787/9789264085442-en.
- CRC Handbook of Chemistry and Physics, (Eds.: W.M. Haynes, D.R. Lide, T.J. Bruno), CRC Press: Boca Raton, FL, 2017. - P. 9–57.