РАЗЛИЧИЯ И СХОДСТВО В СТРАТЕГИЯХ ИСПОЛЬЗОВАНИЯ КОДОНОВ У НЕКОТОРЫХ ФЛАВИВИРУСОВ
Тюлько Ж.С.1, Якименко В.В.2
1кандидат биологических наук, доцент, Омский государственный медицинский университет, 2 доктор биологических наук, старший научный сотрудник, Омский НИИ природно-очаговых инфекций
РАЗЛИЧИЯ И СХОДСТВО В СТРАТЕГИЯХ ИСПОЛЬЗОВАНИЯ КОДОНОВ У НЕКОТОРЫХ ФЛАВИВИРУСОВ
Аннотация
В статье приведены результаты сравнительного изучения стратегий кодирования аминокислотных последовательностей некоторых флавивирусов семейства Flaviviridae по значениям показателей RSCUk, рассчитанных для кодирующей части геномов. Значения RSCUk анализировались методами дискриминантного анализа. По рассчитанным значениям дискриминантных функций для каждой последовательности, были построены диаграммы рассеяния. Они продемонстрировали различия в использовании синонимичных кодонов у разных подтипов вируса клещевого энцефалита, а также внутри группы включающей вирусы: японского энцефалита, денге, Западного Нила, Повассан, леса Кьясанур, желтой лихорадки, клещевого энцефалита, омской геморрагической лихорадки, Тюлений, Лангат, Louping Ill. В результате было показано - различные генотипы вируса клещевого энцефалита не одинаково используют значительную часть синонимичных кодонов, Впервые выявлено также большое сходство в использовании синонимичных кодонов у вирусов Повассан и вирусов омской геморрагической лихорадки.
Ключевые слова: флавивирусы, RSCUk, дискриминантный анализ, стратегия кодирования, вирус клещевого энцефалита, омская геморрагическая лихорадка.
Tyulko J.S.1, Yakimenko V.V.2
1 PhD in Biology, associate professor, Omsk State Medical University, 2 PhD in Biology, head of the Laboratory of arbovirus infections, Omsk Research Institute of Natural Foci Infections
DIFFERENT AND SIMILAR CODON USAGE STRATEGIES IN SOME FLAVIVIRUSES
Abstract
The topic of this paper is a comparative study of the amino acid sequences coding strategies of some of the flavivirus family Flaviviridae. RSCUk values were calculated for the viral genome coding regions. Then RSCUk values were analyzed using discriminant analysis. As a result of the calculated values of the discriminant function for each sequence were constructed scatterplot. Sscatterplots have demonstrated differences in the use of synonymous codons in different subtypes of the tick-borne encephalitis virus as well as within the group consisting of flaviviruses: Japanese encephalitis, dengue, West Nile, Povassan, forests Kyasanur, yellow fever, tick-borne encephalitis virus, Omsk hemorrhagic fever virus, Tyuleniy virus, Langat, Louping Ill. It was demonstrated differences in the use of synonymous codons in different subtypes of the tick-borne encephalitis virus. It revealed a great similarity in the use of synonymous codons in Povassan viruses and virus Omsk hemorrhagic fever.
Key words: flavivirus, RSCUk, discriminant analysis; strategy of protein coding, tick-borne encephalitis virus; Omsk hemorrhagic fever virus.
Введение. К роду Flavivirus семейства Flaviviridae относят более 70 вирусов, сгруппированных в несколько антигенных групп, большинство которых являются арбовирусами, передающимися клещами или комарами, хотя некоторые могут передаваться и теми и другими (вирус Западного Нила), а для некоторых переносчики не обнаружены [8]. Флавивирусные инфекции это природные зоонозы, имеющие большое ветеринарное и медицинское значение, они зачастую вызывают заболевания, плохо поддающиеся лечению и приводящие к нетрудоспособности [2,5].
Флавивирусный вирион (диаметр ≈ 40-50 нм) состоит из нуклеокпсида и липопротеиновой оболочки, покрывающей его. Нуклеокапсид вирусной частицы флавивирусов, включающий в себя капсидный белок С и геномную +РНК, окружен липидной мембраной, в которую включены М –мембранный (8 кДа) и E – оболочечный (50 кДА) белки. Геномная РНК флавивирусов является инфекционной (≈ 11 тыс. нуклеиновых оснований, далее - н.о.), кодирует три структурных (С, М и Е) и семь неструктурных белков (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5), последовательно считываемых в единой рамке считывания и необходимых для размножения вируса в клетках хозяина. Репликация геномной РНК происходит в цитоплазме по полуконсервативному механизму, инициация трансляции осуществляется по кэпзависимому механизму [8]. Для большинства флавивирусов существует общая схема расположения регуляторных элементов РНК, сохраняющихся структур РНК, консервативных и вариабельных участков генома [4,14,20]. Известно также, что вторичная структура РНК очень чувствительна к возникновению мутаций. Эффекты, вызываемые появлением синонимичных или не синонимичных замен, определяются местом их возникновения в геноме, которое может соответствовать положению регуляторных элементов РНК, а также особенностями вторичной структуры РНК, измененной их появлением.
Поэтому, в последнее время все большее внимание привлекают сравнительные исследования влияния особенностей изменчивости нуклеотидной последовательности вирусов на их свойства. При этом интерес представляет не только изучение эффектов, возникающих от замен в конкретных участках генома разных вирусов [3,4], но и общий анализ статистических свойств вирусных нуклеотидных последовательностей [11]. В связи с накоплением в банках данных значительного количества полноразмерных геномов многих флавивирусов, стало возможным провести работы по исследованию смещения в их нуклеотидном составе [11,15]. В этих работах было показано повышенное содержание нуклеотидов C и G, которое считается признаком отбора, в пользу термодинамически устойчивых молекул, а также показано наличие различий по этому показателю для флавивирусов в том числе и ВКЭ [6,11,15,19], что свидетельствует о существовании отбора и некой стратегии в использовании синонимичных кодонов. Поэтому интерес представляло исследование различий в использовании кодонов у флавивирусов, имеющих достаточное представление полногеномными кодирующими последовательностями в генетических банках данных.
Цель исследования: сравнение стратегий использования кодонов у различных флавивирусов по полноразмерным кодирующим нуклеотидным последовательностям их РНК.
Материалы и методы. Для анализа использовались полноразмерные кодирующие последовательности флавивирусов из GenBank, доступные на 2014 г.: японского энцефалита (ЯЭ, 224 последовательности), денге (3512), Западного Нила (ЗН; 613), Повассан (15), леса Кьясанур (28), желтой лихорадки (ЖЛ; 44), клещевого энцефалита (КЭ; 114), омской геморрагической лихорадки (ОГЛ; 5), Тюлений (5), Лангат (4), Louping Ill (4).
Для всех кодирующих последовательностей рассчитывались показатели относительного использования синонимичных кодонов, обозначаемые как RSCUk (Relative Synonymous Codon Usage), для каждого кодона k (стоп кодоны и кодоны, кодирующиеся только одним триплетом, не учитывались). Показатель RSCUk применяют для проведения корректных сравнений частот использования синонимичных кодонов в различных сериях [1,16,17,18]. Он оценивает неслучайность появления кодона k при кодировании аминокислоты, а также позволяет сравнить схемы кодирования в разных генах. Большие значения RSCUk соответствуют более частому использованию кодона. Показатели RSCUk были получены с помощью программ, созданных на базе пакета статистического анализа R. Далее, значения показателей RSCUk для всех типов кодонов нуклеотидной последовательности каждого вируса анализировались с помощью модуля «Общее модели дискриминантного анализа» (general discriminant analysis) программы STATISTICA 6. Полученные по результатам дискриминантного анализа обучающей выборки, значения дискриминантных функций, позволяют построить диаграммы рассеяния этих значений, наглядно представляющие различия в использовании синонимичных кодонов у разных видов флавивирусов, а также проводить классификацию последовательностей, не входивших в обучающую выборку.
Результаты и обсуждение. Первоначально этот метод анализа гомологичных вирусных нуклеотидных последовательностей был применен нами для исследования вируса КЭ, представителя семейства РНК-вирусов Flaviviridae, который эндемичен для многих стран Евразии, где периодически наблюдается заметный рост заболеваемости. Наш анализ подтверждает политипичность вирусов КЭ, который разделяют на три основных подтипа (1-дальневосточный, 2-европейский и 3-сибирский), каждый из которых характеризуется широким ареалом, и двух подтипов (4- тип 178_79 и 5- тип 886_84), локально распространенных в Прибайкалье [2,12]. В рамках каждого подтипа, за последние годы, выделено несколько геновариантов вируса, и вероятно, дальнейшие исследования увеличат их число. Это можно предполагать, так как, максимальное различие между кодирующими нуклеотидными последовательностями разных подтипов, первоначально оцениваемое в 5-6% [12], по мере выявления новых геновариантов вируса КЭ увеличилось до 20%. На основании чего, был поставлен вопрос о квалификации основных подтипов в качестве самостоятельных видов вирусов [2]. Было показано также, что в пределах одного подтипа наблюдается большее сходство в использовании синонимичных кодонов, чем между разными подтипами вирусов КЭ, независимо от способа изоляции штамма вируса, что подтверждает данные, полученные нами ранее [9]. На рисунке 1 дана диаграмма рассеяния для значений дискриминантных функций, рассчитанных для каждой полноразмерной последовательности ВКЭ. Видно, что все случаи хорошо разделяются на три группы, в соответствии с тремя генотипами.
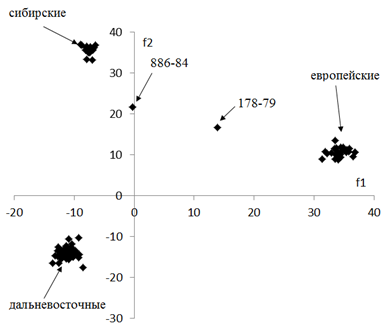
Рис. 1 - Диаграмма рассеяния значений дискриминантных функций, рассчитанных для кодирующих последовательностей вирусов КЭ. По оси абсцисс отложены значения дискриминантной функции f1, по оси ординат - f2. Каждый маркер на схеме соответствует последовательности из банка данных, для которой были рассчитаны значения функций.
Дискриминантный анализ дает возможность изучать различия между двумя и более группами объектов (нуклеотидных последовательностей) по нескольким переменным (RSCUk) одновременно [7,10], а также интерпретировать межгрупповые различия и определять вклад каждой переменной при классификации объектов. При построении диаграммы мы не закладывали никаких эволюционных моделей, анализировались только частотные характеристики использования синонимичных кодонов (RSCUk), а не их расположение в последовательностях. Однако разбиение на кластеры в полученной схеме соответствует разделению вирусов на подтипы, получаемому при выполнении филогенетических построений, учитывающих места возникновения замен. Исходя из этого, можно предположить, что отбор конкретных синонимичных кодонов может быть важной частью процесса микроэволюции ВКЭ, который отражается в структуре филогении вируса и поэтому требует дальнейшего изучения, как механизм, возможно приводящий к формированию новых подтипов вирусов КЭ, на первых шагах изменяя преимущественно нуклеотидный состав генома, а впоследствии приводя к появлению новых аминокислотных последовательностей отличающихся от исходных. Такое предположение поддерживает характеристику, которая ранее была дана подтипу 4 « ...штамм 178-79, имеющий собственный, отличный от других генотипов вируса КЭ набор нуклеотидов в составе изученного фрагмента РНК, по аминокислотной последовательности идентичен генотипу 1» [2].
Примечательно, что использование тех же самых кодонов оставалось значимым для классификации последовательностей, как при сравнительном анализе различных подтипов КЭ, так и при анализе отдельных геновариантов в рамках одного и того же подтипа (были проанализированы дальневосточный и сибирский подтипы).
Результаты, дискриминантного анализа значений RSCUk для полноразмерных кодирующих последовательностей вируса КЭ, подтвердили хорошую применимость данного метода для исследования флавивирусов, и наш дальнейший анализ различий в использовании синонимичных кодонов включил в себя флавивирусы ЯЭ, денге, ЗН, Повассан, леса Кьясанур, ЖЛ, КЭ, ОГЛ, Тюлений, Лангат, Louping Ill.
На рисунке 2 представлена схема полученной классификации. Как и ожидалось, из-за возможного пассирования вируса в разных биологических системах при лабораторных исследованиях, не показано однозначного соответствия схем использования синонимичных кодонов конкретному хозяину. Тем не менее, выявляется определенное сходство в использовании синонимичных триплетов у представителей разных флавивирусов использующих схожих хозяев (только комаров или только клещей).
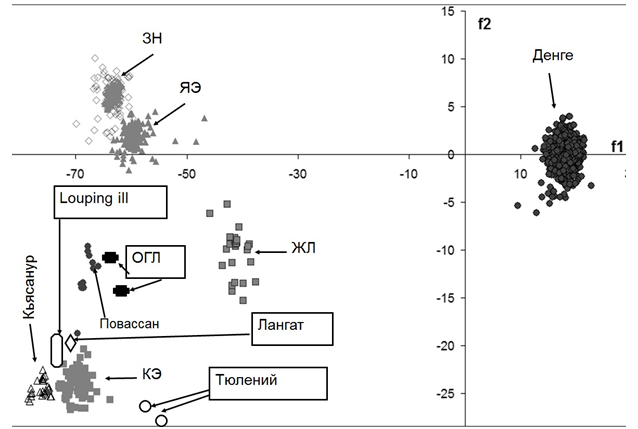
Рис. 2 - Диаграмма рассеяния значений дискриминантных функций, рассчитанных для кодирующих последовательностей флавивирусов. По оси абсцисс отложены значения дискриминантной функции f1, по оси ординат - f2. Каждому виду вирусов соответствует определенная форма маркера, каждый маркер на схеме соответствует последовательности из банка данных, для которой были рассчитаны значения функций. Названия, обведенные рамкой, указывают на флавивирусные последовательности, не включенные в обучающую выборку, но классифицированные с помощью функций f1 и f2, полученных при ее анализе. Их расположение соответствует расположению остальных вирусов из группы клещевых энцефалитов.
Полученный результат отражает, как действие отбора, проявляющееся на аминокислотном уровне, так и наличие определенных стратегий кодирования, приводящих к различному частотному использованию синонимичных кодонов разными группами вирусов без анализа причин их возникновения. Данный подход позволил выявить сходство в стратегиях кодирования у вирусов Повассан и ОГЛ, а также Кьясанур и КЭ, в то время как филогенетические построения и исследования смещения в нуклеотидном составе для этих вирусов не предоставляют подобной информации [13, 19]. Молекулярный механизм, формирующий это сходство, может быть достаточно сложным и включать в себя популяционные взаимодействия с определенным кругом хозяев, характерных для каждого вида.
Механизмы меж- и внутривидовой дивергенции вирусов не всегда очевидны и достаточно трудны для понимания и обнаружения. В случае РНК-геномных вирусов, их изменчивость определяется не только естественным отбором, действующим на формирование аминокислотных последовательностей вирусных белков (возникновение несинонимичных нуклеотидных замен), но и существованием структурных требований, задаваемых вторичной и третичной структурами вирусной РНК (возникновение как синонимичных, так и несинонимичных нуклеотидных замен). Поэтому, важное значение имеет изучение закономерностей возникновения в геноме вируса синонимичных нуклеотидных замен. Подобные закономерности невозможно правильно оценить без привлечения многомерных статистических методов и автоматизации анализа, проводимого для большого числа геномных последовательностей вируса. Сейчас, эти исследования стали возможными и необходимыми вследствие значительного увеличения количества секвенированных нуклеотидных последовательностей флавивирусов. Наиболее подходящими для этого являются полногеномные последовательности.
Литература
- Бутвиловский А.В. Изучение стратегии кодирования белков / А.В.Бутвиловский, В.Э.Бутвиловский, Е.А.Черноус // Медицинский журнал. -2009. –T.2. -С.24-7.
- Вотяков В.И. Клещевые энцефалиты Евразии (вопросы экологии, молекулярной эпидемиологии, нозоологии, эволюции) / В.И.Вотяков, В.И.Злобин, Н.П.Мишаева. – Новосибирск: Наука, 2002. – 438 с.
- Демина Т.В. Вопросы генотипирования и анализ генетической вариабельности вируса клещевого энцефалита: Автореф. дисс. докт. биол. наук. - Иркутск, 2013. - 46 с.
- Карганова Г.Г. Хозяин-специфические детерминанты в геноме вируса клещевого энцефалита. // В кн.: Фундаментальные и прикладные аспекты изучения паразитических членистоногих в XXI в. СПб.: -2013. -С. 71-3.
- Леонова Г.Н. Клещевой энцефалит в Приморском крае. Владивосток: Дальнаука, 1997. -190 с.
- Лукашев В.В. Молекулярная эволюция и филогенетический анализ / В.В. Лукашев. -М.: БИНОМ, -2009. -256 с.
- Орлов А.И. Прикладная статистика: учебник. М.: Экзамен; 2006.
- Руководство по вирусологии: Вирусы и вирусные инфекции человека и животных/ Под.ред. Д.К. Львова.- М.: ООО «Издательство «Медицинское информационное агентство», 2013. - 1200 с.
- Тюлько Ж.С., Якименко В.В. Оценка внутривидовой изменчивости при анализе использования кодонов у различных подтипов ВКЭ // Национальные приоритеты России Материалы научной конференции с международным участием, посвященной 75-летию теории академика Е.Н. Павловского о природной очаговости болезней. Омск, -2014. № 3, -С. 117 –120.
- Халафян А.А. Учебник STATISTIKA Статистический анализ данных. М.: Бином-Пресс, -2007.- 512 c.
- Belalov I.S. Causes and Implications of Codon Usage Bias in RNA Viruses / I.S. Belalov, A.N. Lukashev // PLOS ONE. -2013; -Vol.8, №2. [Электронный ресурс] URL: http://www.plosone.org/article/info%3Adoi%2F1371%2Fjournal.pone.0056642. (дата обращения 11.04.2015).
- Ecker M. Sequence analysis and genetic classification of tick-borne encephalitis viruses from Europe and Asia / M. Ecker, S.L. Allison, T. Meixner, F.X. Heinz // Journal of General Virology — 1999. - Vol.80. -P.179–185.
- Grard G. Genetic characterization of tick-borne flaviviruses: New insights into evolution, pathogenetic determinants and taxonomy / G. Grard, G. Moureau, R.N. Charrel J. Lemasson, J. Gonzalez, P. Gallian, T.S. Gritsun, E.C. Holmes, E.A. Gould, X. de Lamballerie // Virology. -2007. -Vol.361. –P.80–92.
- Gould E.A., De Lamballerie X., De Zanotto A., Holmes E.C. Flavivirus evolution // Proceedings of the 3th International conference "Ticks and tick-borne Pathogens: into the 21st century", Bratislava. - – P. 1 – 9.
- Moosavi F. Analysis of synonymous codon usage bias and nucleotide and amino acid composition in 13 species of Flaviviridae / F. Moosavi, H.Mohabatkar, S. Mohsenzadeh //Journal of Cell and Molecular Research. -2011. -Vol.3, №1. -P.1-11.
- Perriere G. Use and misuse of correspondence analysis in codon usage studies/ G.Perriere, Thioulouse // Nucleic Acids Research. - 2002. -Vol. 30, №20. -P. 4548-55.
- Qian W. Balanced Codon Usage Optimizes Eukaryotic Translational Efficiency./W.Qian, J-R.Yang, N.M.Pearson, C.Maclean, J.Zhang //PLoS Genet. -2012. -Vol.8, №3. [Электронный ресурс] URL: http://www.plosgenetics.org/article/info%3Adoi%2F10.1371%2Fjournal.pgen.1002603. (дата обращения 11.04.2015).
- Sharp P. M. Codon usage in yeast cluster-analysis clearly differentiates highly and lowly expressed genes / P.M.Sharp, T.M.F.Tuohy, K.R.Mosurski // Nucleic Acids Research. -1986. -Vol.14. -P.5125- 5143.
- Schubert A.M. Evolution of the sequence composition of Flaviviruses. / A.M.Schubert, C.Putonti // Infection, Genetics and Evolution. -2010. -Vol.10, №1. -P.129-36.
- Thurner C. Conserved RNA secondary structures in Flaviviridae genomes/ C. Thurner, Witwer, I.L. Hofacker, P.F. Stadler // Journal of General Virology. – 2004. -Vol. 85. – P.1113–1124.
References
- Butvilovskij A.V. Izuchenie strategii kodirovanija belkov / A.V.Butvilovskij, V.Je.Butvilovskij, E.A.Chernous // Medicinskij zhurnal. -2009. –T.2. -S.24-7.
- Votjakov V.I. Kleshhevye jencefality Evrazii (voprosy jekologii, molekuljarnoj jepidemiologii, nozoologii, jevoljucii) / V.I.Votjakov, V.I.Zlobin, N.P.Mishaeva. – Novosibirsk: Nauka, 2002. – 438 s.
- Demina T.V. Voprosy genotipirovanija i analiz geneticheskoj variabel'nosti virusa kleshhevogo jencefalita: Avtoref. diss. dokt. biol. nauk. - Irkutsk, 2013. - 46 s.
- Karganova G.G. Hozjain-specificheskie determinanty v genome virusa kleshhevogo jencefalita. // V kn.: Fundamental'nye i prikladnye aspekty izuchenija paraziticheskih chlenistonogih v XXI v. SPb.: -2013. -S. 71-3.
- Leonova G.N. Kleshhevoj jencefalit v Primorskom krae. Vladivostok: Dal'nauka, 1997. -190 s.
- Lukashev V.V. Molekuljarnaja jevoljucija i filogeneticheskij analiz / V.V. Lukashev. -M.: BINOM, -2009. -256 s.
- Orlov A.I. Prikladnaja statistika: uchebnik. : Jekzamen; 2006.
- Rukovodstvo po virusologii: Virusy i virusnye infekcii cheloveka i zhivotnyh/ Pod.red. D.K. L'vova.- M.: OOO «Izdatel'stvo «Medicinskoe informacionnoe agentstvo», 2013. - 1200 s.
- Tjulko J.S., Jakimenko V.V. Ocenka vnutrividovoj izmenchivosti pri analize ispol'zovanija kodonov u razlichnyh podtipov VKJe // Nacional'nye prioritety Rossii Materialy nauchnoj konferencii s mezhdunarodnym uchastiem, posvjashhennoj 75-letiju teorii akademika E.N. Pavlovskogo o prirodnoj ochagovosti boleznej. Omsk, -2014. № 3, -S. 117 –120.
- Halafjan A.A. Uchebnik STATISTIKA 6. Statisticheskij analiz dannyh. M.: Binom-Press, -2007.- 512 c.
- Belalov I.S. Causes and Implications of Codon Usage Bias in RNA Viruses / I.S. Belalov, A.N. Lukashev // PLOS ONE. -2013; -Vol.8, №2. [Jelektronnyj resurs] URL: http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0056642. (data obrashhenija 04.2015).
- Ecker M. Sequence analysis and genetic classification of tick-borne encephalitis viruses from Europe and Asia / M. Ecker, S.L. Allison, T. Meixner, F.X. Heinz // Journal of General Virology — 1999. - Vol.80. -P.179–185.
- Grard G. Genetic characterization of tick-borne flaviviruses: New insights into evolution, pathogenetic determinants and taxonomy / G. Grard, G. Moureau, R.N. Charrel J. Lemasson, J. Gonzalez, P. Gallian, T.S. Gritsun, E.C. Holmes, E.A. Gould, X. de Lamballerie // Virology. -2007. -Vol.361. –P.80–92.
- Gould E.A., De Lamballerie X., De Zanotto A., Holmes E.C. Flavivirus evolution // Proceedings of the 3th International conference "Ticks and tick-borne Pathogens: into the 21st century", Bratislava. - – P. 1 – 9.
- Moosavi F. Analysis of synonymous codon usage bias and nucleotide and amino acid composition in 13 species of Flaviviridae / F. Moosavi, H.Mohabatkar, S. Mohsenzadeh //Journal of Cell and Molecular Research. -2011. -Vol.3, №1. -P.1-11.
- Perriere G. Use and misuse of correspondence analysis in codon usage studies/ G.Perriere, Thioulouse // Nucleic Acids Research. - 2002. -Vol. 30, №20. -P. 4548-55.
- Qian W. Balanced Codon Usage Optimizes Eukaryotic Translational Efficiency./W.Qian, J-R.Yang, N.M.Pearson, C.Maclean, J.Zhang //PLoS Genet. -2012. -Vol.8, №3. [Jelektronnyj resurs] URL: http://www.plosgenetics.org/article/info%3Adoi%2F10.1371%2Fjournal.pgen.1002603. (data obrashhenija 04.2015).
- Sharp P. M. Codon usage in yeast cluster-analysis clearly differentiates highly and lowly expressed genes / P.M.Sharp, T.M.F.Tuohy, K.R.Mosurski // Nucleic Acids Research. -1986. -Vol.14. -P.5125- 5143.
- Schubert A.M. Evolution of the sequence composition of Flaviviruses. / A.M.Schubert, C.Putonti // Infection, Genetics and Evolution. -2010. -Vol.10, №1. -P.129-36.
- Thurner C. Conserved RNA secondary structures in Flaviviridae genomes/ C. Thurner, Witwer, I.L. Hofacker, P.F. Stadler // Journal of General Virology. – 2004. -Vol. 85. – P.1113–1124.